

Genome-wide profiling of mouse RNA secondary structures reveals key features of the mammalian transcriptome
- PMID: 25323333
- PMCID: PMC4220049
- DOI: 10.1186/s13059-014-0491-2
Genome-wide profiling of mouse RNA secondary structures reveals key features of the mammalian transcriptome
Abstract
Background: The understanding of RNA structure is a key feature toward the comprehension of RNA functions and mechanisms of action. In particular, non-coding RNAs are thought to exert their functions by specific secondary structures, but an efficient annotation on a large scale of these structures is still missing.
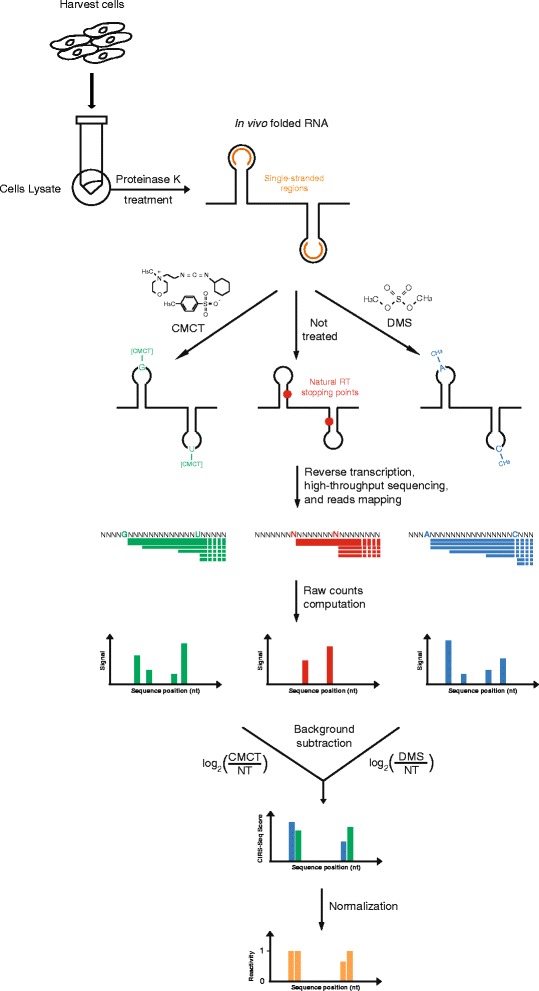
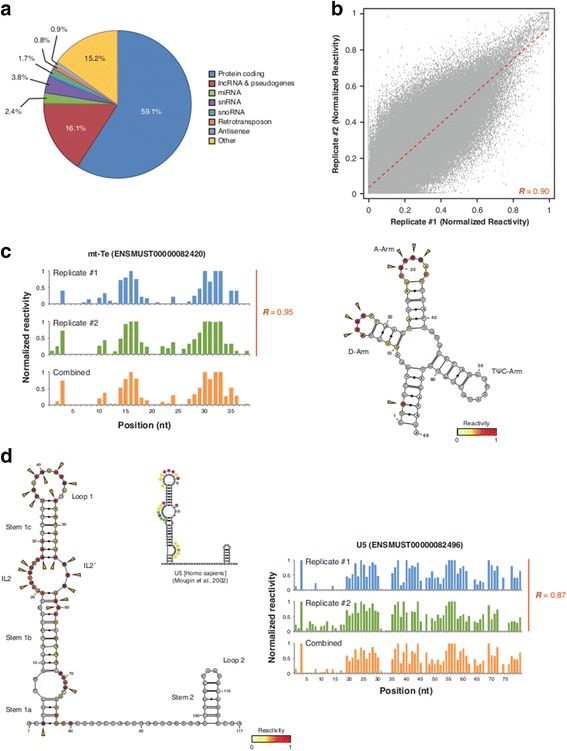
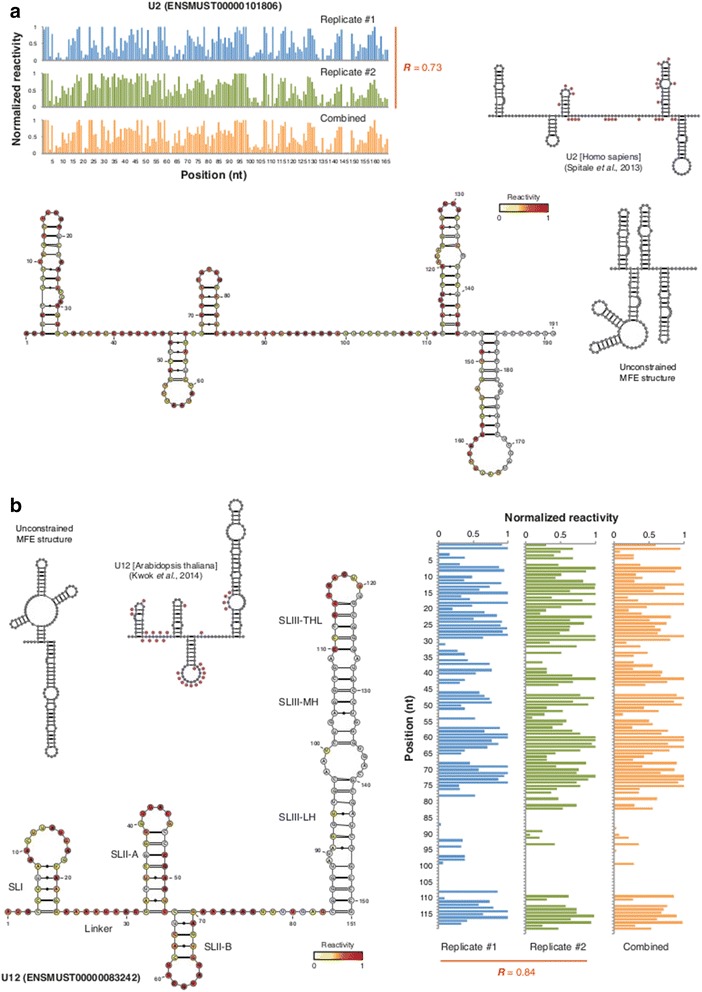
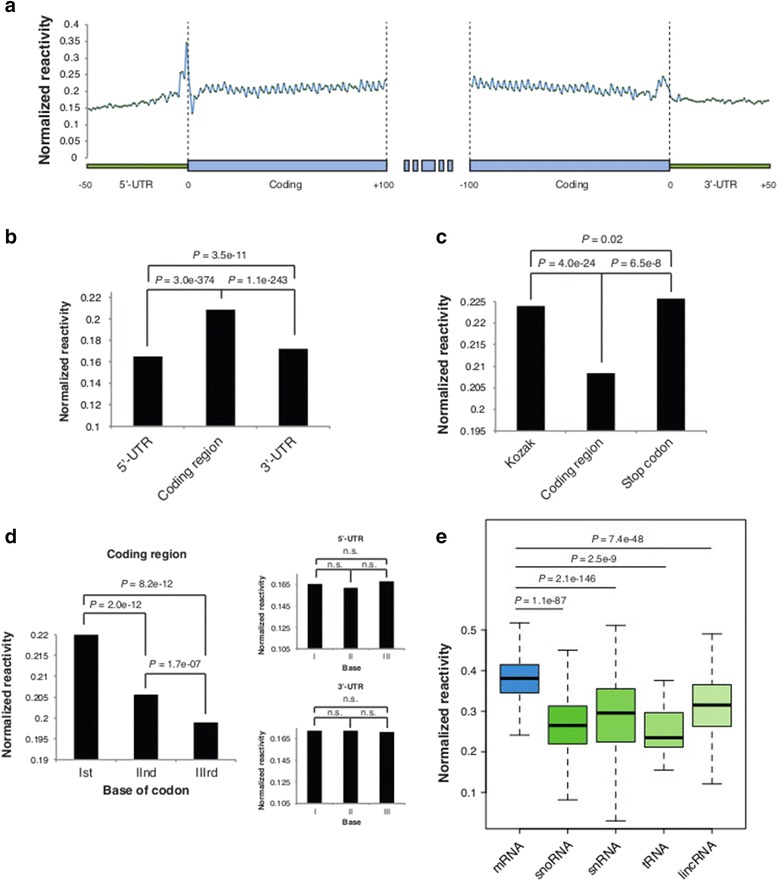
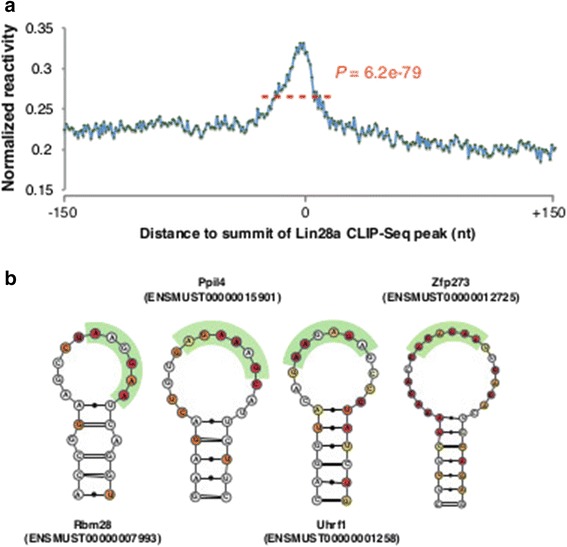
Results: By using a novel high-throughput method, named chemical inference of RNA structures, CIRS-seq, that uses dimethyl sulfate, and N-cyclohexyl- N'-(2-morpholinoethyl) carbodiimide metho-p-toluenesulfonate to modify RNA residues in single-stranded conformation within native deproteinized RNA secondary structures, we investigate the structural features of mouse embryonic stem cell transcripts. Our analysis reveals an unexpected higher structuring of the 5′ and 3′ untranslated regions compared to the coding regions, a reduced structuring at the Kozak sequence and stop codon, and a three-nucleotide periodicity across the coding region of messenger RNAs. We also observe that ncRNAs exhibit a higher degree of structuring with respect to protein coding transcripts. Moreover, we find that the Lin28a binding protein binds selectively to RNA motifs with a strong preference toward a single stranded conformation.
Conclusions: This work defines for the first time the complete RNA structurome of mouse embryonic stem cells,revealing an extremely distinct RNA structural landscape. These results demonstrate that CIRS-seq constitutes an important tool for the identification of native deproteinized RNA structures.
Figures
References
-
- Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, Xue C, Marinov GK, Khatun J, Williams BA, Zaleski C, Rozowsky J, Röder M, Kokocinski F, Abdelhamid RF, Alioto T, Antoshechkin I, Baer MT, Bar NS, Batut P, Bell K, Bell I, Chakrabortty S, Chen X, Chrast J, Curado J, et al. Landscape of transcription in human cells. Nature. 2012;489:101–108. doi: 10.1038/nature11233. - DOI - PMC - PubMed
-
- Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, Guernec G, Martin D, Merkel A, Knowles DG, Lagarde J, Veeravalli L, Ruan X, Ruan Y, Lassmann T, Carninci P, Brown JB, Lipovich L, Gonzalez JM, Thomas M, Davis CA, Shiekhattar R, Gingeras TR, Hubbard TJ, Notredame C, Harrow J, Guigó R. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012;22:1775–1789. doi: 10.1101/gr.132159.111. - DOI - PMC - PubMed
-
- Guttman M, Donaghey J, Carey BW, Garber M, Grenier JK, Munson G, Young G, Lucas AB, Ach R, Bruhn L, Yang X, Amit I, Meissner A, Regev A, Rinn JL, Root DE, Lander ES. lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature. 2011;477:295–300. doi: 10.1038/nature10398. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
