

A rise in NAD precursor nicotinamide mononucleotide (NMN) after injury promotes axon degeneration
- PMID: 25323584
- PMCID: PMC4392071
- DOI: 10.1038/cdd.2014.164
A rise in NAD precursor nicotinamide mononucleotide (NMN) after injury promotes axon degeneration
Abstract
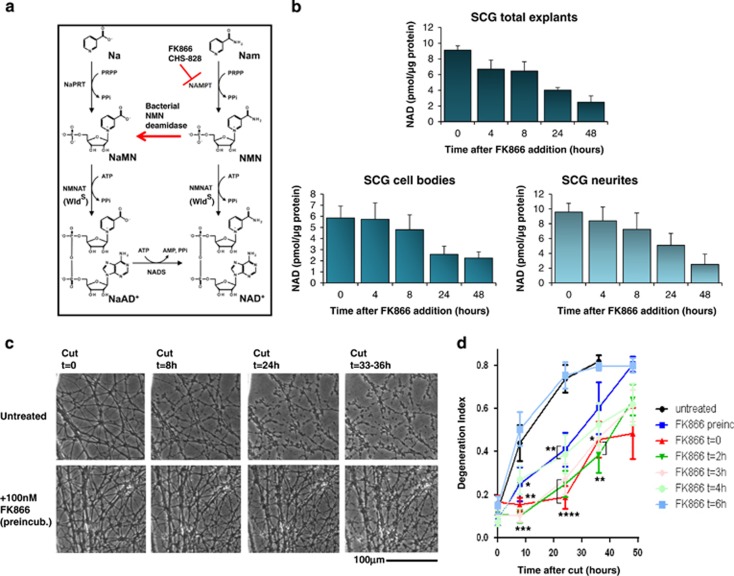
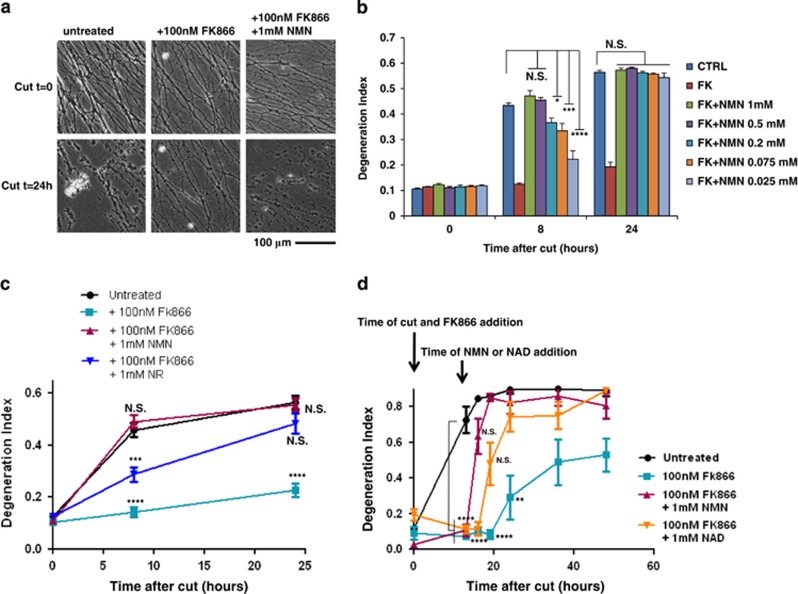
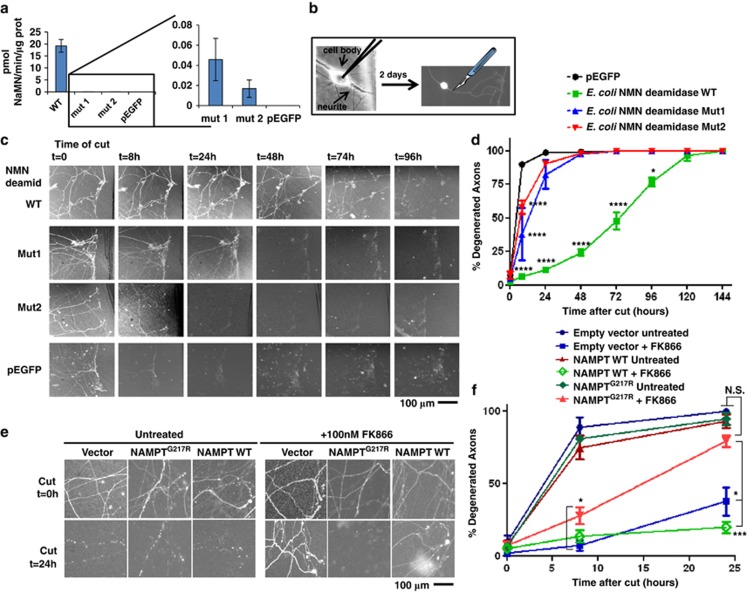
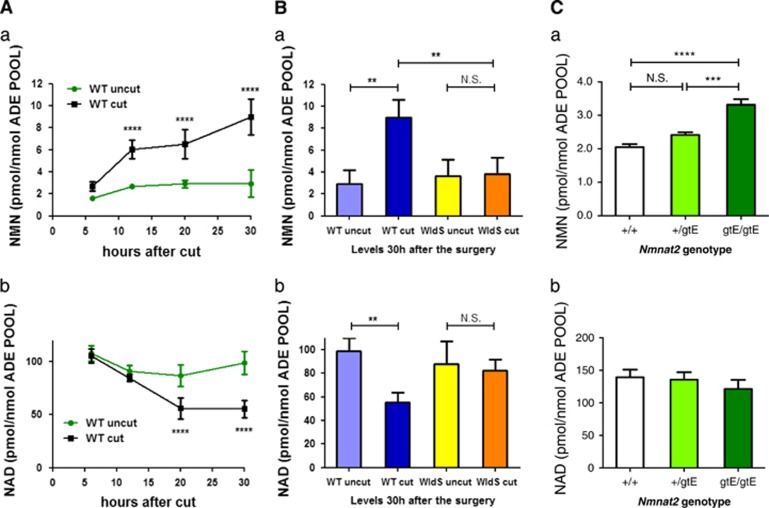
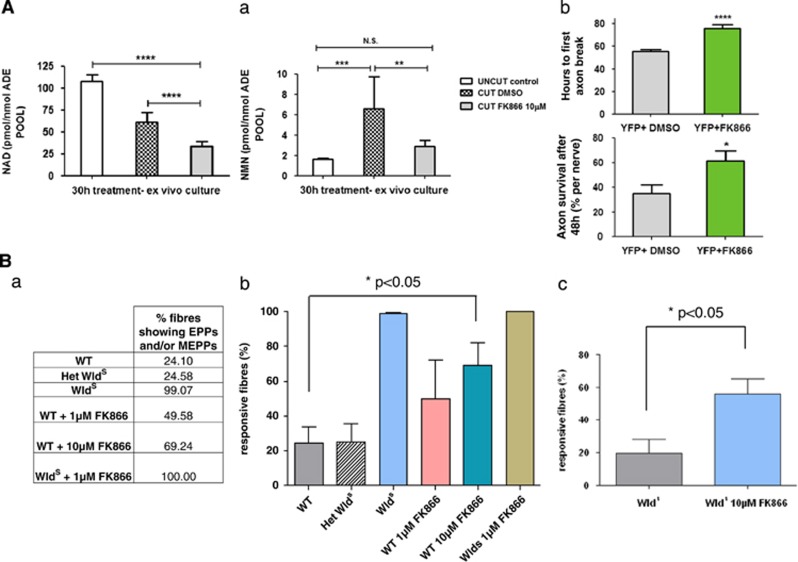
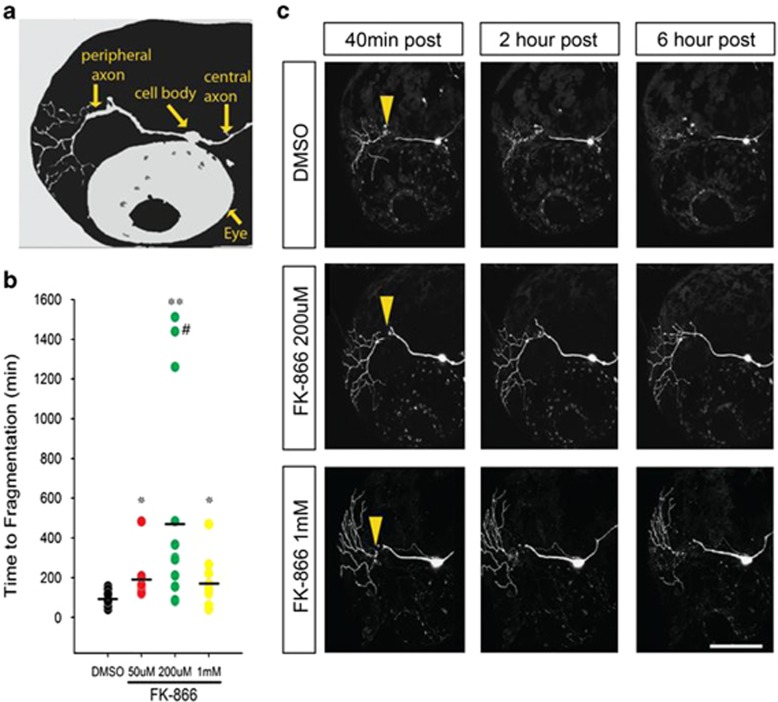
NAD metabolism regulates diverse biological processes, including ageing, circadian rhythm and axon survival. Axons depend on the activity of the central enzyme in NAD biosynthesis, nicotinamide mononucleotide adenylyltransferase 2 (NMNAT2), for their maintenance and degenerate rapidly when this activity is lost. However, whether axon survival is regulated by the supply of NAD or by another action of this enzyme remains unclear. Here we show that the nucleotide precursor of NAD, nicotinamide mononucleotide (NMN), accumulates after nerve injury and promotes axon degeneration. Inhibitors of NMN-synthesising enzyme NAMPT confer robust morphological and functional protection of injured axons and synapses despite lowering NAD. Exogenous NMN abolishes this protection, suggesting that NMN accumulation within axons after NMNAT2 degradation could promote degeneration. Ectopic expression of NMN deamidase, a bacterial NMN-scavenging enzyme, prolongs survival of injured axons, providing genetic evidence to support such a mechanism. NMN rises prior to degeneration and both the NAMPT inhibitor FK866 and the axon protective protein Wld(S) prevent this rise. These data indicate that the mechanism by which NMNAT and the related Wld(S) protein promote axon survival is by limiting NMN accumulation. They indicate a novel physiological function for NMN in mammals and reveal an unexpected link between new strategies for cancer chemotherapy and the treatment of axonopathies.
Figures
References
-
- Stoll G, Jander S, Myers RR. Degeneration and regeneration of the peripheral nervous system: from Augustus Waller's observations to neuroinflammation. J Peripher Nerv Syst. 2002;7:13–27. - PubMed
-
- Lunn ER, Perry VH, Brown MC, Rosen H, Gordon S. Absence of Wallerian degeneration does not hinder regeneration in peripheral nerve. Eur J Neurosci. 1989;1:27–33. - PubMed
-
- Conforti L, Gilley J, Coleman MP. Wallerian degeneration: an emerging axon death pathway linking injury and disease. Nat Rev Neurosci. 2014;15:394–409. - PubMed
-
- Agostini M, Bedin C, Pucciarelli S, Enzo M, Briarava M, Seraglia R, et al. APC I1307K mutations and forkhead box gene (FOXO1A): another piece of an interesting correlation. Int J Biol Markers. 2012;27:13–19. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
