

Clinical characteristics and current therapies for inherited retinal degenerations
- PMID: 25324231
- PMCID: PMC4315917
- DOI: 10.1101/cshperspect.a017111
Clinical characteristics and current therapies for inherited retinal degenerations
Abstract
Inherited retinal degenerations (IRDs) encompass a large group of clinically and genetically heterogeneous diseases that affect approximately 1 in 3000 people (>2 million people worldwide) (Bessant DA, Ali RR, Bhattacharya SS. 2001. Molecular genetics and prospects for therapy of the inherited retinal dystrophies. Curr Opin Genet Dev 11: 307-316.). IRDs may be inherited as Mendelian traits or through mitochondrial DNA, and may affect the entire retina (e.g., rod-cone dystrophy, also known as retinitis pigmentosa, cone dystrophy, cone-rod dystrophy, choroideremia, Usher syndrome, and Bardet-Bidel syndrome) or be restricted to the macula (e.g., Stargardt disease, Best disease, and Sorsby fundus dystrophy), ultimately leading to blindness. IRDs are a major cause of severe vision loss, with profound impact on patients and society. Although IRDs remain untreatable today, significant progress toward therapeutic strategies for IRDs has marked the past two decades. This progress has been based on better understanding of the pathophysiological pathways of these diseases and on technological advances.
Copyright © 2015 Cold Spring Harbor Laboratory Press; all rights reserved.
Figures
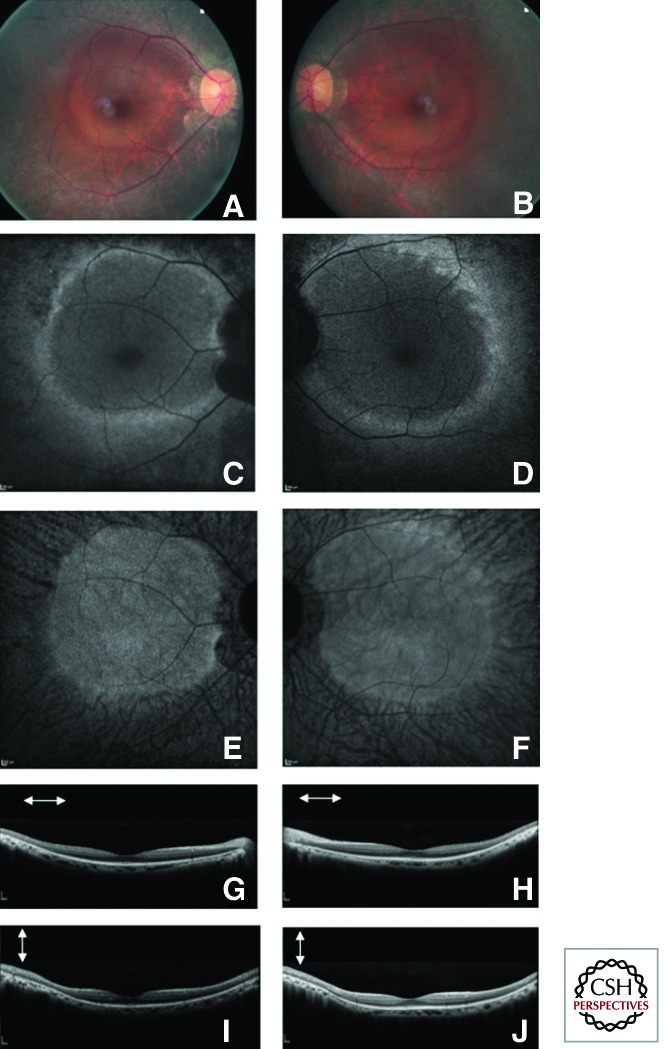
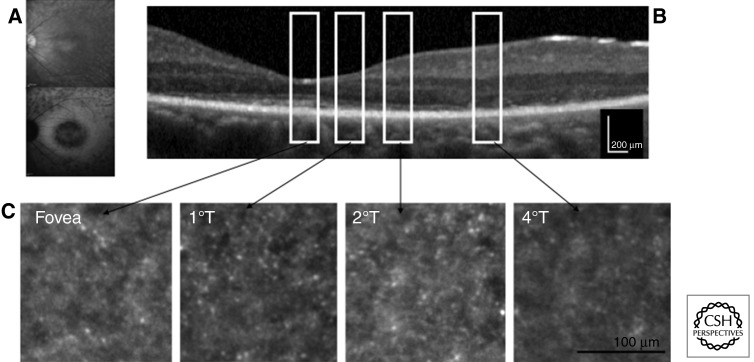
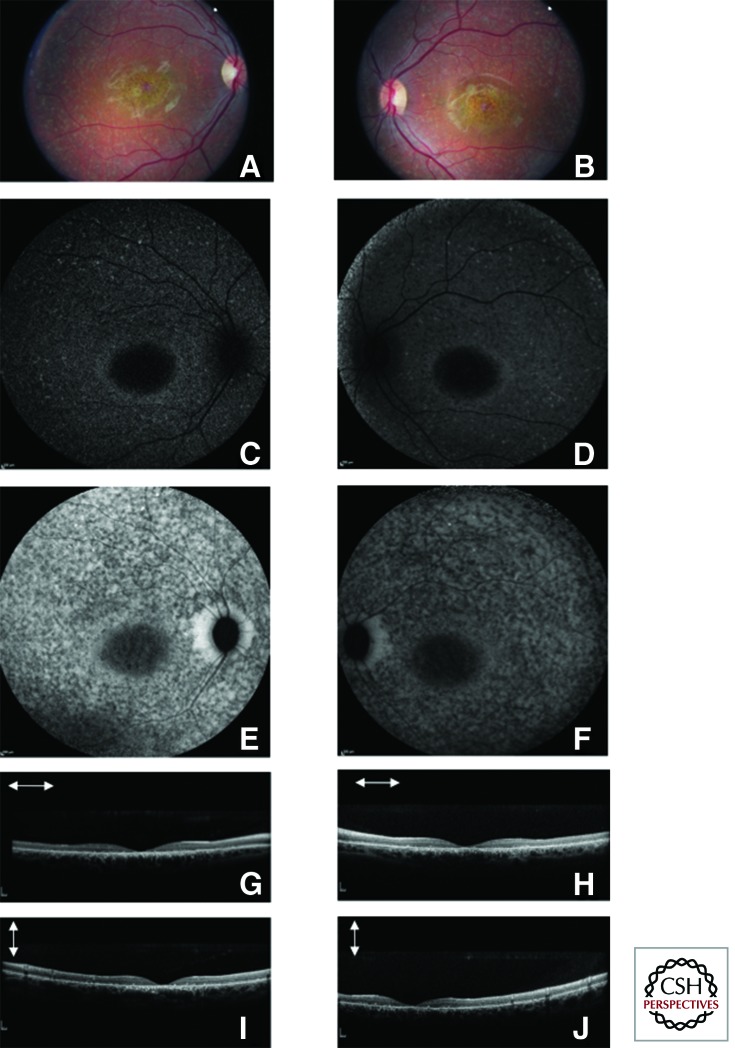
References
-
- Acland GM, Aguirre GD, Ray J, Zhang Q, Aleman TS, Cideciyan AV, Pearce-Kelling SE, Anand V, Zeng Y, Maguire AM, et al. 2001. Gene therapy restores vision in a canine model of childhood blindness. Nat Genet 28: 92–95. - PubMed
-
- Acland GM, Aguirre GD, Bennett J, Aleman TS, Cideciyan AV, Bennicelli J, Dejneka NS, Pearce-Kelling SE, Maguire AM, Palczewski K, et al. 2005. Long-term restoration of rod and cone vision by single dose rAAV-mediated gene transfer to the retina in a canine model of childhood blindness. Mol Ther 12: 1072–1082. - PMC - PubMed
-
- Adackapara CA, Sunness JS, Dibernardo CW, Melia BM, Dagnelie G 2008. Prevalence of cystoid macular edema and stability in oct retinal thickness in eyes with retinitis pigmentosa during a 48-week lutein trial. Retina 28: 103–110. - PubMed
-
- Aleman TS, Duncan JL, Bieber ML, de Castro E, Marks DA, Gardner LM, Steinberg JD, Cideciyan AV, Maguire MG, Jacobson SG 2001. Macular pigment and lutein supplementation in retinitis pigmentosa and Usher syndrome. Invest Ophthalmol Vis Sci 42: 1873–1881. - PubMed
-
- Ali RR, Sarra GM, Stephens C, Alwis MD, Bainbridge JW, Munro PM, Fauser S, Reichel MB, Kinnon C, Hunt DM, et al. 2000. Restoration of photoreceptor ultrastructure and function in retinal degeneration slow mice by gene therapy. Nat Genet 25: 306–310. - PubMed
Publication types
MeSH terms
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous