

Ubiquitin-proteasome system involvement in Huntington's disease
- PMID: 25324717
- PMCID: PMC4179678
- DOI: 10.3389/fnmol.2014.00077
Ubiquitin-proteasome system involvement in Huntington's disease
Abstract
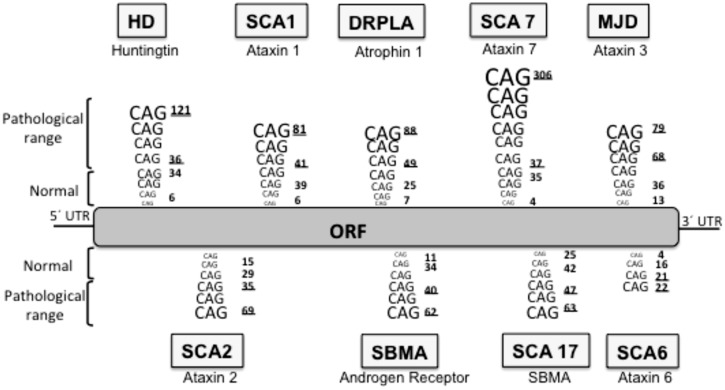
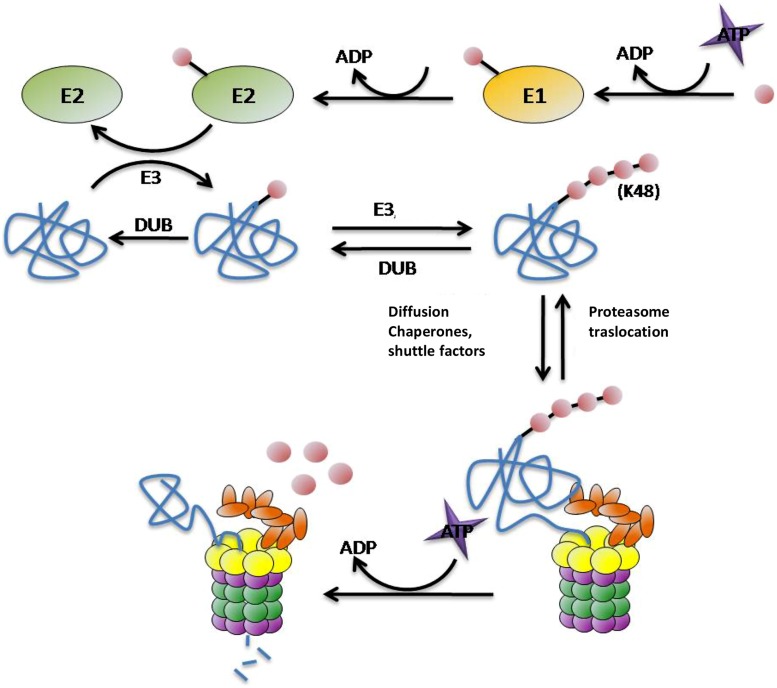
Huntington's disease (HD) is a genetic autosomal dominant neurodegenerative disease caused by the expansion of a CAG repeat in the huntingtin (htt) gene. This triplet expansion encodes a polyglutamine stretch (polyQ) in the N-terminus of the high molecular weight (348-kDa) and ubiquitously expressed protein htt. Normal individuals have between 6 and 35 CAG triplets, while expansions longer than 40 repeats lead to HD. The onset and severity of the disease depend on the length of the polyQ tract: the longer the polyglutamine stretch (polyQ) is, the earlier the disease begins and the more severe the symptoms are. One of the main histopathological hallmarks of HD is the presence of intraneuronal proteinaceous inclusion bodies, whose prominent and invariant feature is the presence of ubiquitin (Ub); therefore, they can be detected with anti-ubiquitin and anti-proteasome antibodies. This, together with the observation that mutations in components of the ubiquitin-proteasome system (UPS) give rise to some neurodegenerative diseases, suggests that UPS impairment may be causative of HD. Even though the link between disrupted Ub homeostasis and protein aggregation to HD is undisputed, the functional significance of these correlations and their mechanistic implications remains unresolved. Moreover, there is no consistent evidence documenting an accompanying decrease in levels of free Ub or disruption of Ub pool dynamics in neurodegenerative disease or models thus suggesting that the Ub-conjugate accumulation may be benign and just underlie lesion in 26S function. In this chapter we will elaborate on the different studies that have been performed using different experimental approaches, in order to shed light to this matter.
Keywords: Huntington’s disease; animal models; degron-fluorescent proteins; inclusion body; ubiquitin–proteasome system.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources