

Energy landscapes of functional proteins are inherently risky
- PMID: 25325699
- PMCID: PMC4416114
- DOI: 10.1038/nchembio.1670
Energy landscapes of functional proteins are inherently risky
Abstract
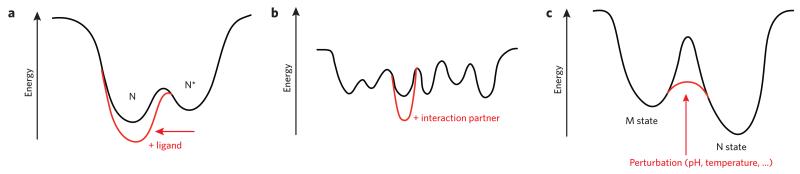
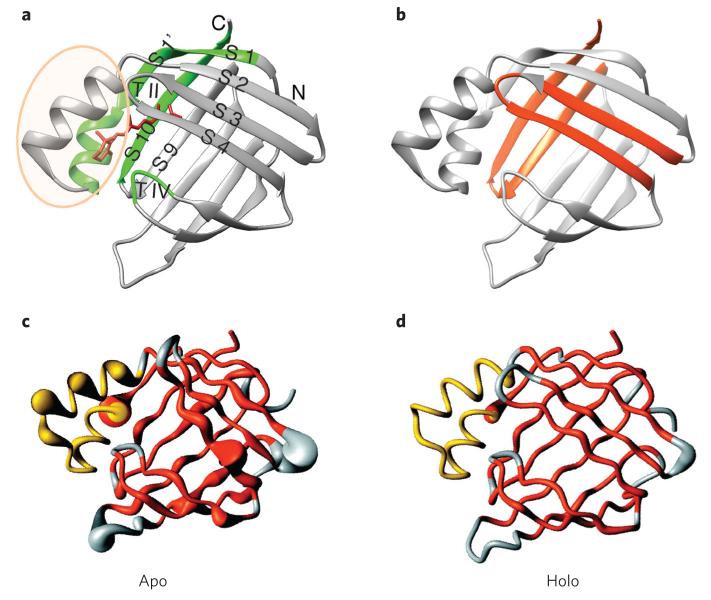
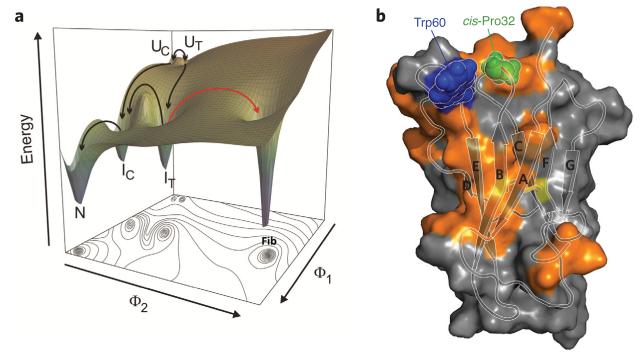
Evolutionary pressure for protein function leads to unavoidable sampling of conformational states that are at risk of misfolding and aggregation. The resulting tension between functional requirements and the risk of misfolding and/or aggregation in the evolution of proteins is becoming more and more apparent. One outcome of this tension is sensitivity to mutation, in which only subtle changes in sequence that may be functionally advantageous can tip the delicate balance toward protein aggregation. Similarly, increasing the concentration of aggregation-prone species by reducing the ability to control protein levels or compromising protein folding capacity engenders increased risk of aggregation and disease. In this Perspective, we describe examples that epitomize the tension between protein functional energy landscapes and aggregation risk. Each case illustrates how the energy landscapes for the at-risk proteins are sculpted to enable them to perform their functions and how the risks of aggregation are minimized under cellular conditions using a variety of compensatory mechanisms.
Figures


References
-
- Knowles TP, Vendruscolo M, Dobson CM. The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 2014;15:384–396. - PubMed
-
- Uversky VN. Intrinsic disorder in proteins associated with neurodegenerative diseases. Front. Biosci. (Landmark Ed.) 2009;14:5188–5238. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- GM027616/GM/NIGMS NIH HHS/United States
- MC_PC_13054/MRC_/Medical Research Council/United Kingdom
- 322408/ERC_/European Research Council/International
- R01 GM094848/GM/NIGMS NIH HHS/United States
- 092896/WT_/Wellcome Trust/United Kingdom
- MC_U117584256/MRC_/Medical Research Council/United Kingdom
- R01 GM027616/GM/NIGMS NIH HHS/United States
- U117584256/MRC_/Medical Research Council/United Kingdom
- WT092896MA/WT_/Wellcome Trust/United Kingdom
- R01 GM101644/GM/NIGMS NIH HHS/United States
- R01 GM060418/GM/NIGMS NIH HHS/United States
- GM060418/GM/NIGMS NIH HHS/United States
- GM101644/GM/NIGMS NIH HHS/United States
- GM094848/GM/NIGMS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
