

In vitro, long-range sequence information for de novo genome assembly via transposase contiguity
- PMID: 25327137
- PMCID: PMC4248320
- DOI: 10.1101/gr.178319.114
In vitro, long-range sequence information for de novo genome assembly via transposase contiguity
Abstract
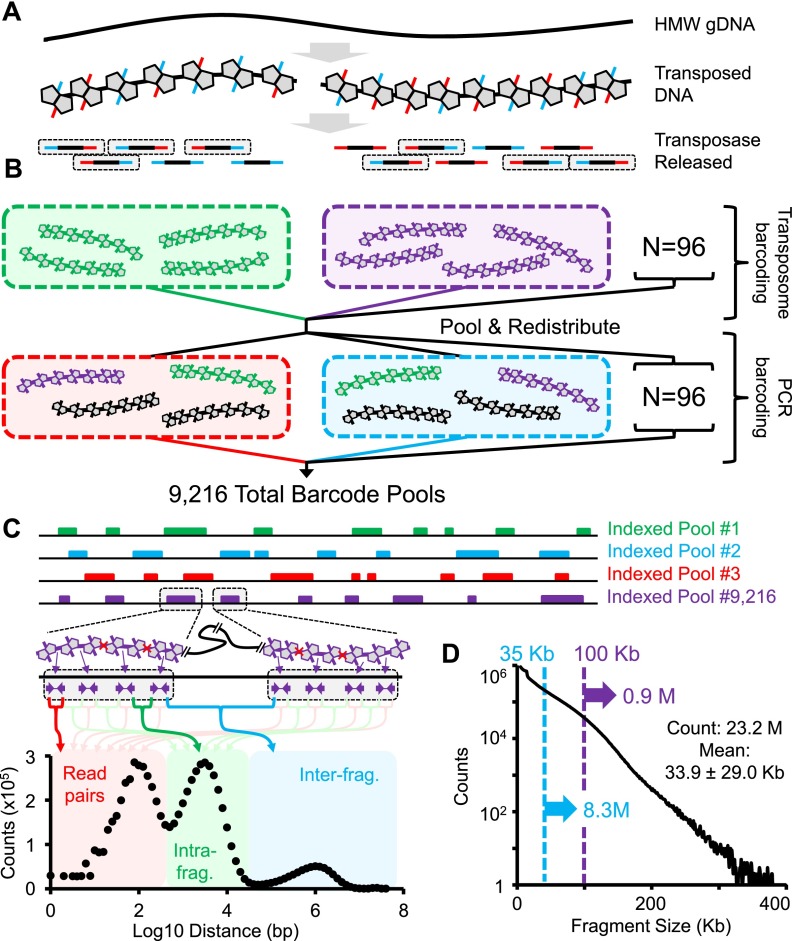
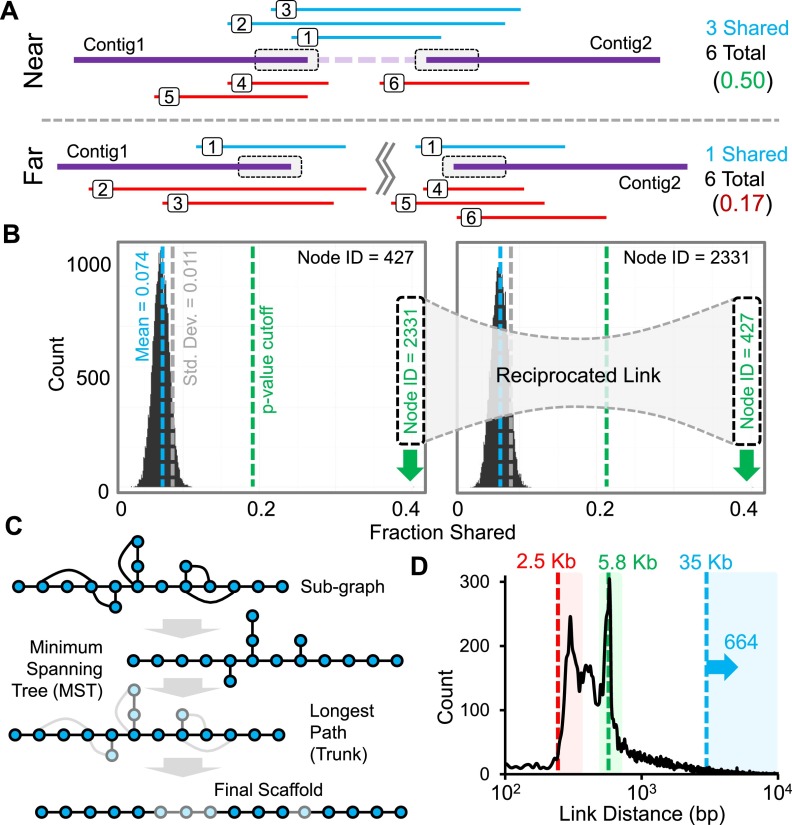
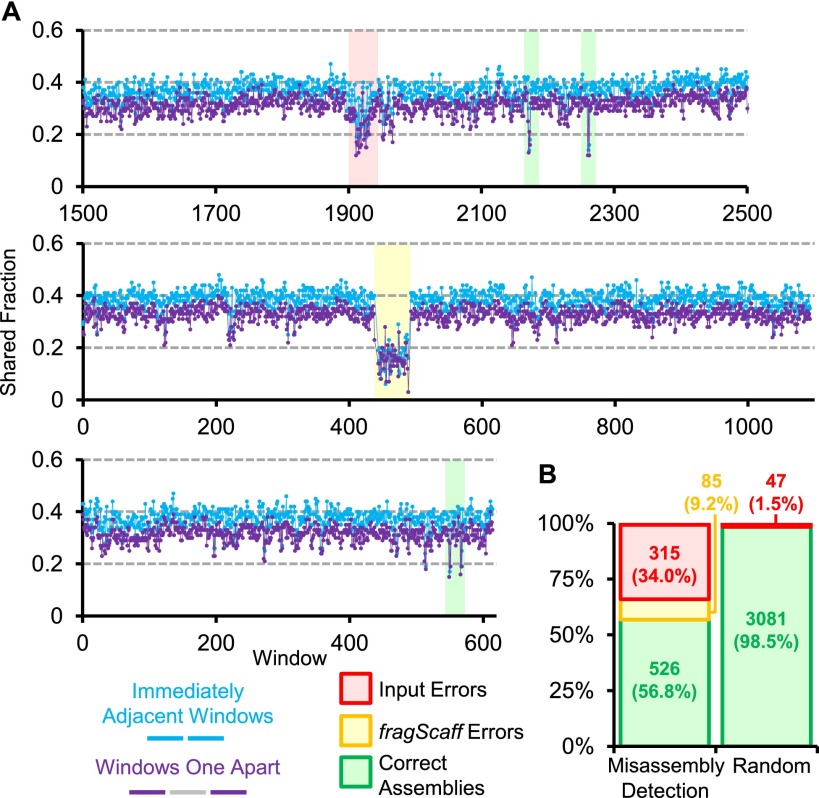
We describe a method that exploits contiguity preserving transposase sequencing (CPT-seq) to facilitate the scaffolding of de novo genome assemblies. CPT-seq is an entirely in vitro means of generating libraries comprised of 9216 indexed pools, each of which contains thousands of sparsely sequenced long fragments ranging from 5 kilobases to > 1 megabase. These pools are "subhaploid," in that the lengths of fragments contained in each pool sums to ∼5% to 10% of the full genome. The scaffolding approach described here, termed fragScaff, leverages coincidences between the content of different pools as a source of contiguity information. Specifically, CPT-seq data is mapped to a de novo genome assembly, followed by the identification of pairs of contigs or scaffolds whose ends disproportionately co-occur in the same indexed pools, consistent with true adjacency in the genome. Such candidate "joins" are used to construct a graph, which is then resolved by a minimum spanning tree. As a proof-of-concept, we apply CPT-seq and fragScaff to substantially boost the contiguity of de novo assemblies of the human, mouse, and fly genomes, increasing the scaffold N50 of de novo assemblies by eight- to 57-fold with high accuracy. We also demonstrate that fragScaff is complementary to Hi-C-based contact probability maps, providing midrange contiguity to support robust, accurate chromosome-scale de novo genome assemblies without the need for laborious in vivo cloning steps. Finally, we demonstrate CPT-seq as a means of anchoring unplaced novel human contigs to the reference genome as well as for detecting misassembled sequences.
© 2014 Adey et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources