

Insulin regulation of myocardial autophagy
- PMID: 25327953
- PMCID: PMC5609463
- DOI: 10.1253/circj.cj-14-1080
Insulin regulation of myocardial autophagy
Abstract
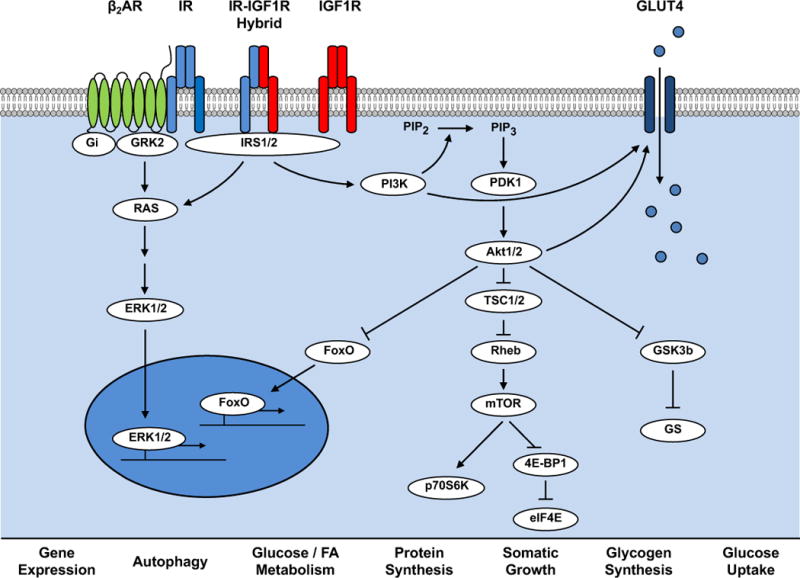
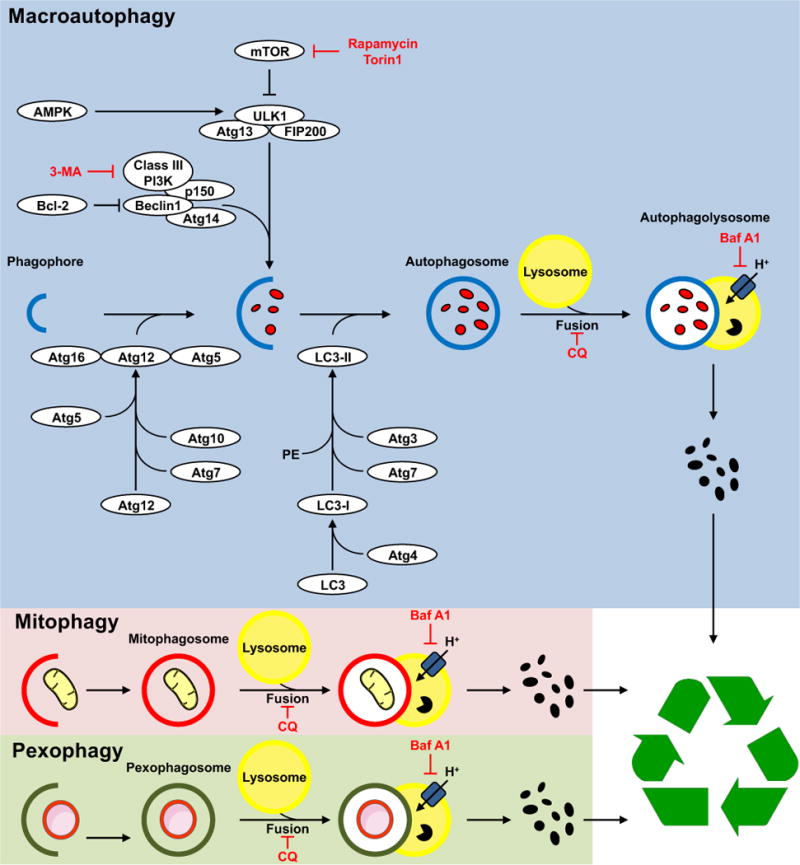
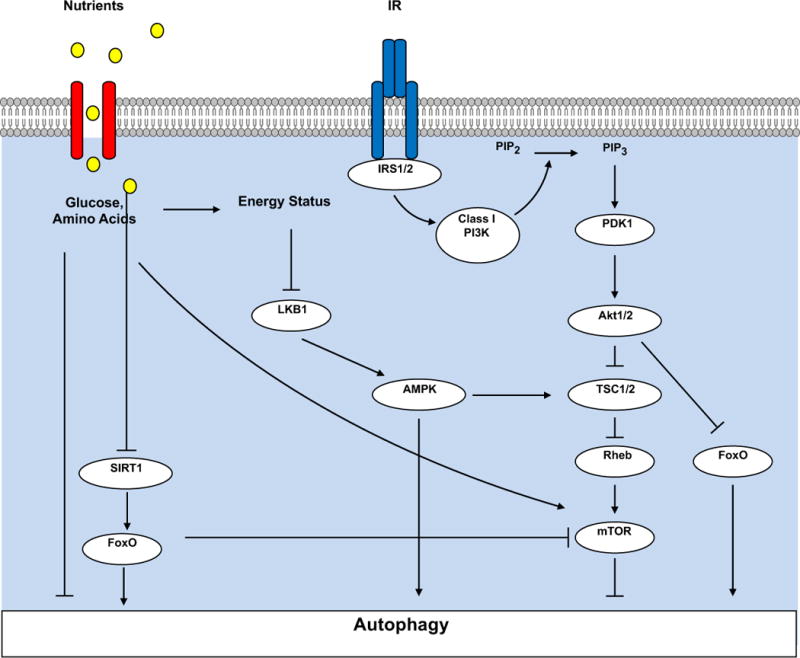
Autophagy is a conserved cellular process that plays an important role in cardiovascular homeostasis. Basal levels of autophagy are required for the maintenance of organellar quality control. Autophagy is dynamically regulated in the heart in the fasting to re-feeding transition. Insulin signaling plays an important role in the regulation of myocardial fuel metabolism, mitochondrial function and cellular growth. Recent studies have suggested an important role for insulin signaling in the regulation of myocardial autophagy. This dynamic regulation of autophagy induction during fasting may contribute to organellar homeostasis and if perturbed under conditions of hyperinsulinemia could contribute to accelerated cardiac aging.
Conflict of interest statement
The authors have no conflicts of interest to declare.
Figures
References
-
- Muniyappa R, Montagnani M, Koh KK, Quon MJ. Cardiovascular actions of insulin. Endocr Rev. 2007;28:463–491. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical