

MacSyFinder: a program to mine genomes for molecular systems with an application to CRISPR-Cas systems
- PMID: 25330359
- PMCID: PMC4201578
- DOI: 10.1371/journal.pone.0110726
MacSyFinder: a program to mine genomes for molecular systems with an application to CRISPR-Cas systems
Abstract
Motivation: Biologists often wish to use their knowledge on a few experimental models of a given molecular system to identify homologs in genomic data. We developed a generic tool for this purpose.
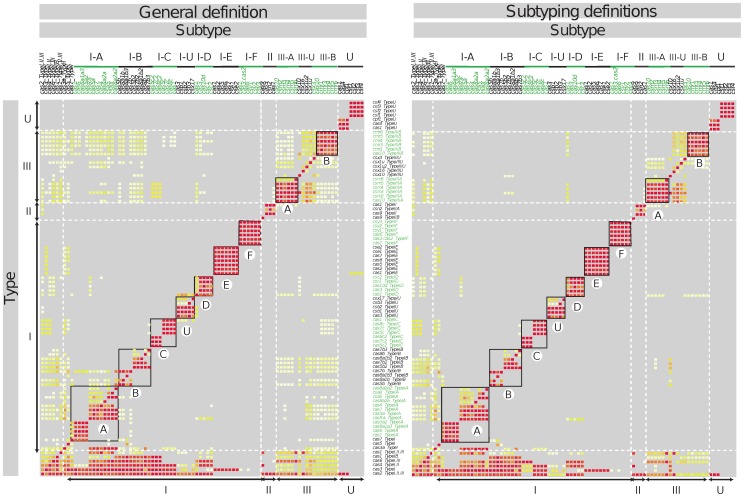
Results: Macromolecular System Finder (MacSyFinder) provides a flexible framework to model the properties of molecular systems (cellular machinery or pathway) including their components, evolutionary associations with other systems and genetic architecture. Modelled features also include functional analogs, and the multiple uses of a same component by different systems. Models are used to search for molecular systems in complete genomes or in unstructured data like metagenomes. The components of the systems are searched by sequence similarity using Hidden Markov model (HMM) protein profiles. The assignment of hits to a given system is decided based on compliance with the content and organization of the system model. A graphical interface, MacSyView, facilitates the analysis of the results by showing overviews of component content and genomic context. To exemplify the use of MacSyFinder we built models to detect and class CRISPR-Cas systems following a previously established classification. We show that MacSyFinder allows to easily define an accurate "Cas-finder" using publicly available protein profiles.
Availability and implementation: MacSyFinder is a standalone application implemented in Python. It requires Python 2.7, Hmmer and makeblastdb (version 2.2.28 or higher). It is freely available with its source code under a GPLv3 license at https://github.com/gem-pasteur/macsyfinder. It is compatible with all platforms supporting Python and Hmmer/makeblastdb. The "Cas-finder" (models and HMM profiles) is distributed as a compressed tarball archive as Supporting Information.
Conflict of interest statement
Figures
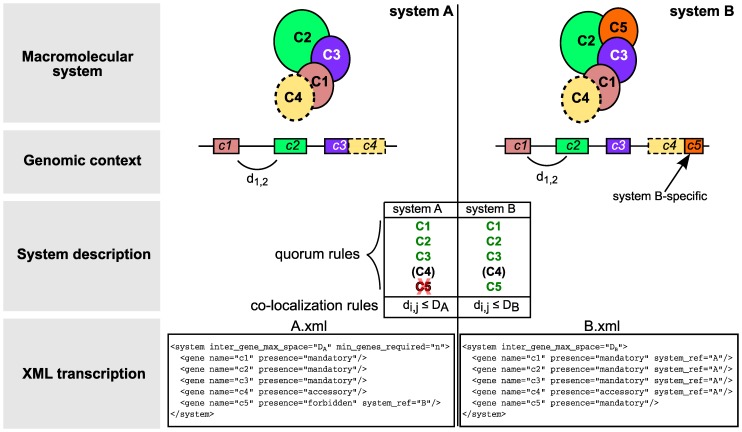
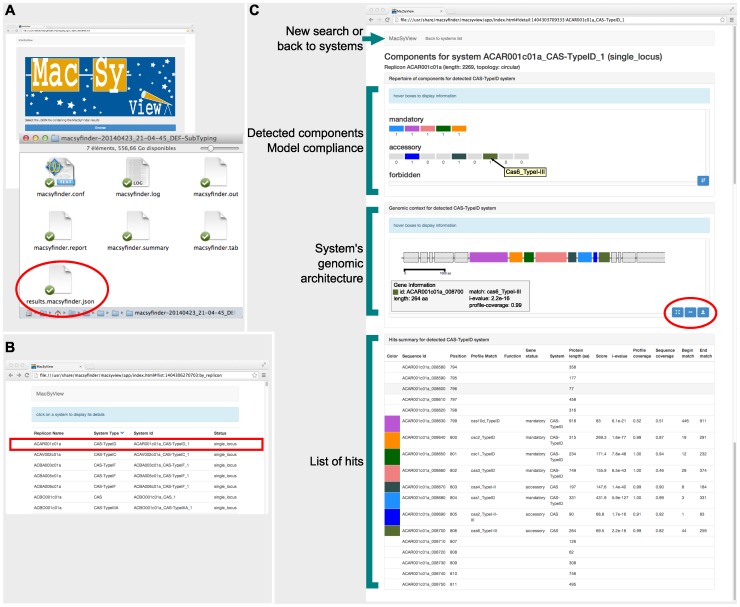
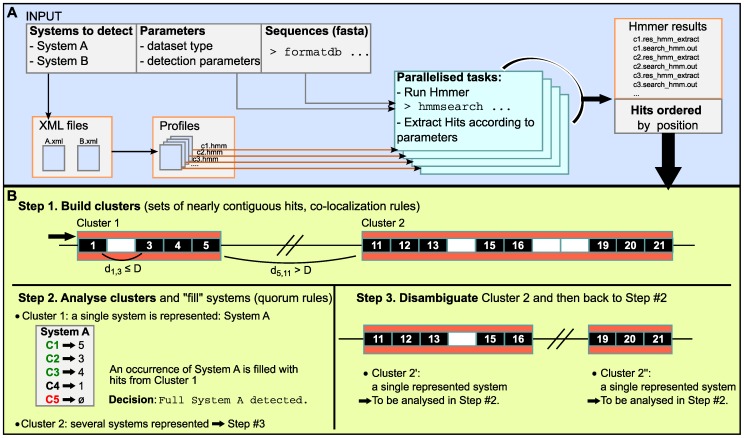
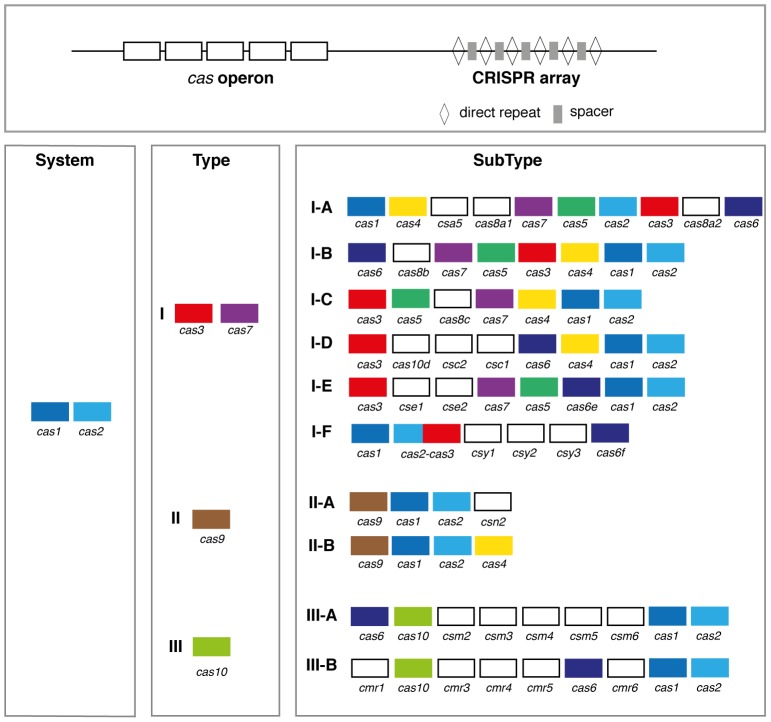
References
-
- Alberts B (1998) The cell as a collection of protein machines: preparing the next generation of molecular biologists. Cell 92: 291–294. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
