

Redox regulation of NF-κB p50 and M1 polarization in microglia
- PMID: 25331559
- PMCID: PMC4322433
- DOI: 10.1002/glia.22762
Redox regulation of NF-κB p50 and M1 polarization in microglia
Abstract
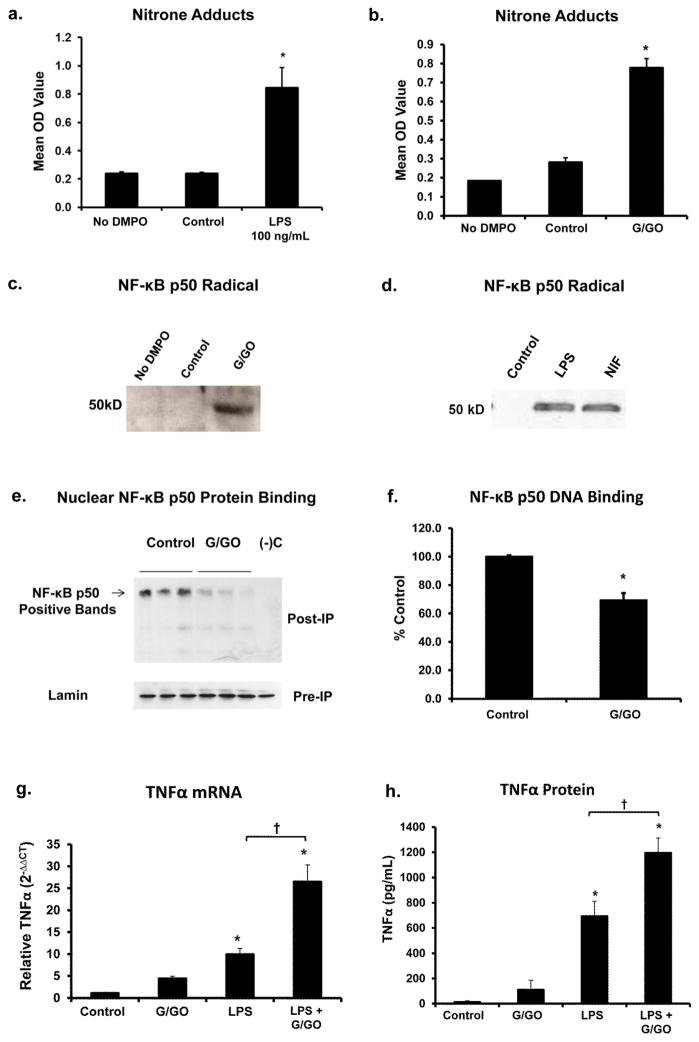
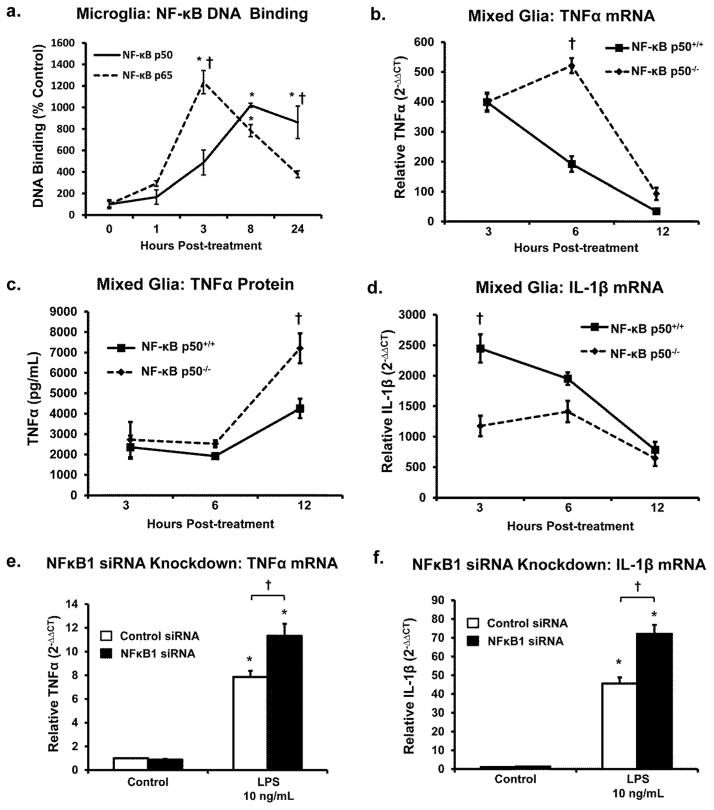
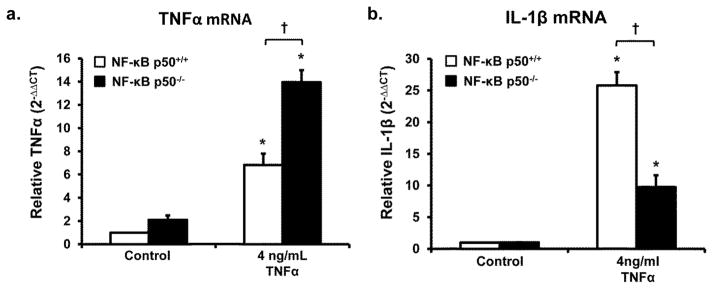
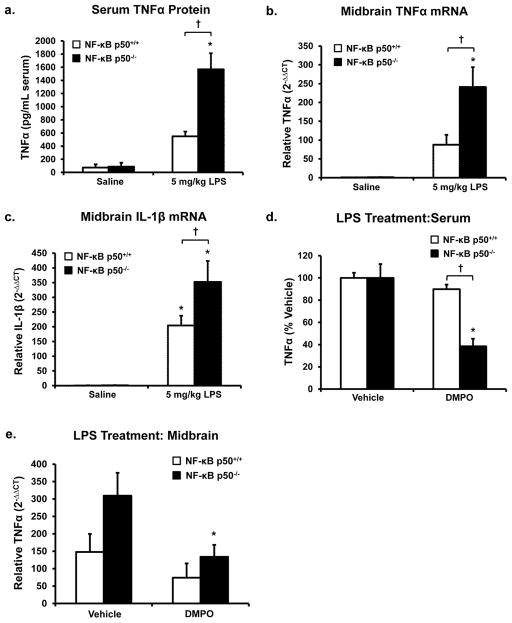
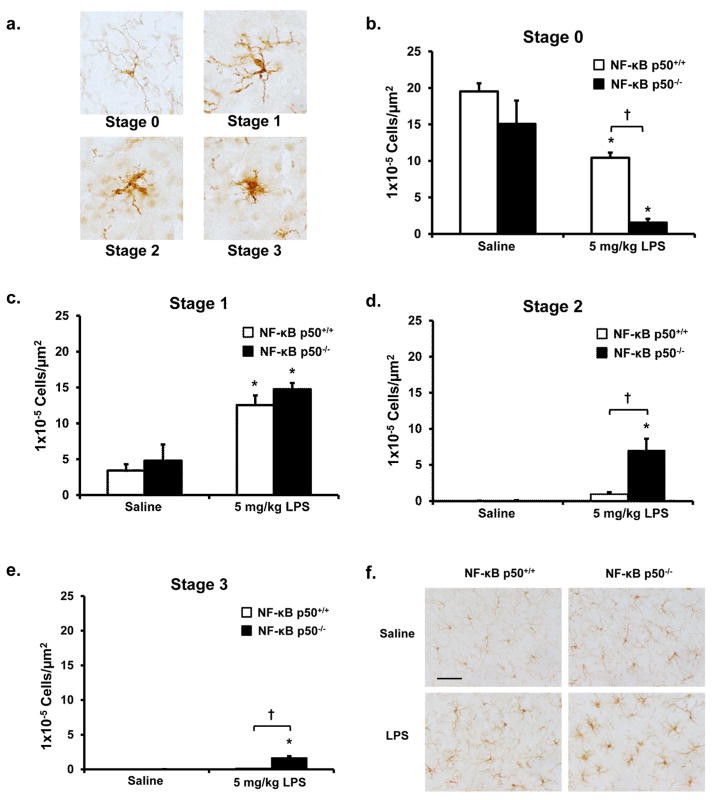
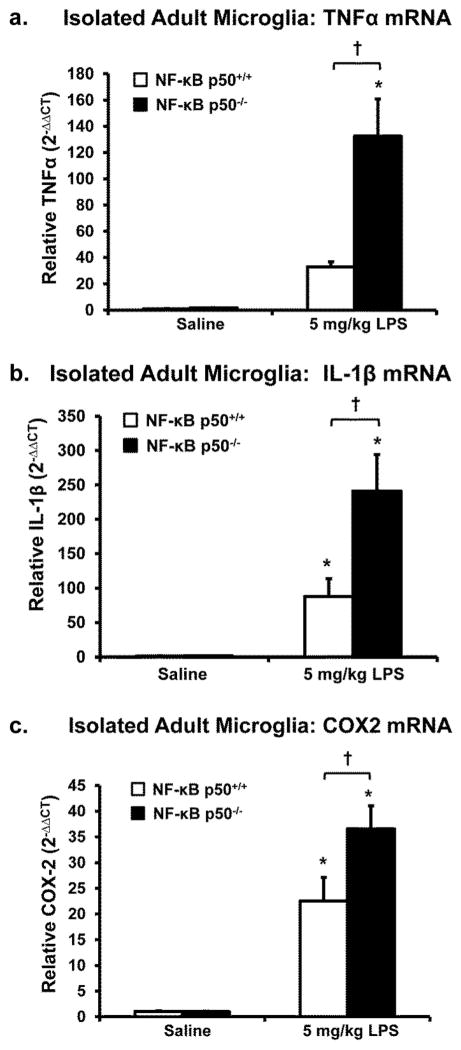
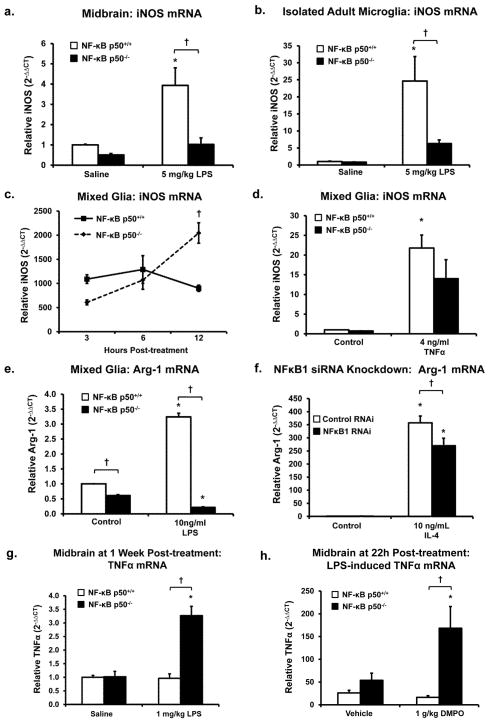
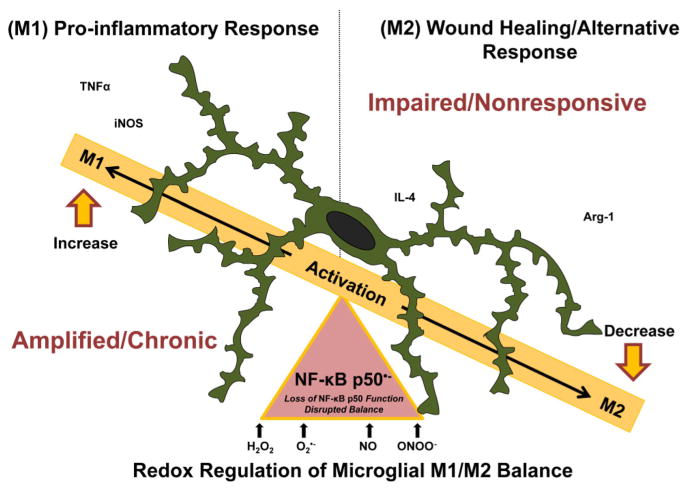
Redox-signaling is implicated in deleterious microglial activation underlying CNS disease, but how ROS program aberrant microglial function is unknown. Here, the oxidation of NF-κB p50 to a free radical intermediate is identified as a marker of dysfunctional M1 (pro-inflammatory) polarization in microglia. Microglia exposed to steady fluxes of H2 O2 showed altered NF-κB p50 protein-protein interactions, decreased NF-κB p50 DNA binding, and augmented late-stage TNFα expression, indicating that H2 O2 impairs NF-κB p50 function and prolongs amplified M1 activation. NF-κB p50(-/-) mice and cultures exhibited a disrupted M2 (alternative) response and impaired resolution of the M1 response. Persistent neuroinflammation continued 1 week after LPS (1 mg/kg, IP) administration in the NF-κB p50(-/-) mice. However, peripheral inflammation had already resolved in both strains of mice. Treatment with the spin-trap DMPO mildly reduced LPS-induced 22 h TNFα in the brain in NF-κB p50(+/+) mice. Interestingly, DMPO failed to reduce and strongly augmented brain TNFα production in NF-κB p50(-/-) mice, implicating a fundamental role for NF-κB p50 in the regulation of chronic neuroinflammation by free radicals. These data identify NF-κB p50 as a key redox-signaling mechanism regulating the M1/M2 balance in microglia, where loss of function leads to a CNS-specific vulnerability to chronic inflammation.
Keywords: CNS; H2O2; NF-κB p50; microglia; redox signaling.
© 2014 Wiley Periodicals, Inc.
Conflict of interest statement
The authors have no competing interests to declare.
Figures
References
-
- Block ML, Wu X, Pei Z, Li G, Wang T, Qin L, Wilson B, Yang J, Hong JS, Veronesi B. Nanometer size diesel exhaust particles are selectively toxic to dopaminergic neurons: the role of microglia, phagocytosis, and NADPH oxidase. FASEB J. 2004;18:1618–20. - PubMed
-
- Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
