

Fibrodysplasia ossificans progressiva: clinical course, genetic mutations and genotype-phenotype correlation
- PMID: 25337067
- PMCID: PMC4188166
- DOI: 10.1159/000365770
Fibrodysplasia ossificans progressiva: clinical course, genetic mutations and genotype-phenotype correlation
Abstract
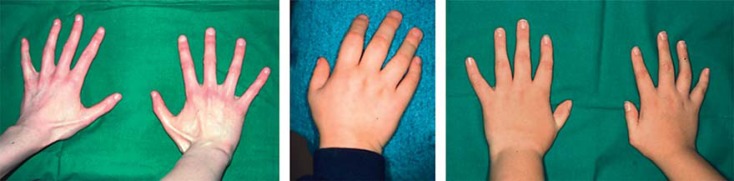
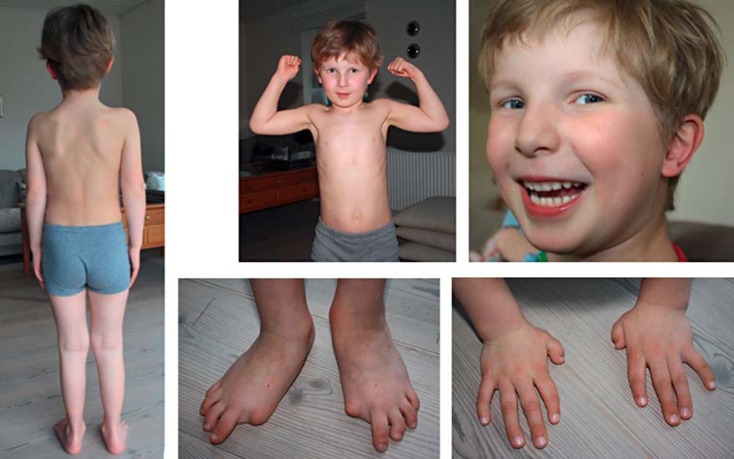
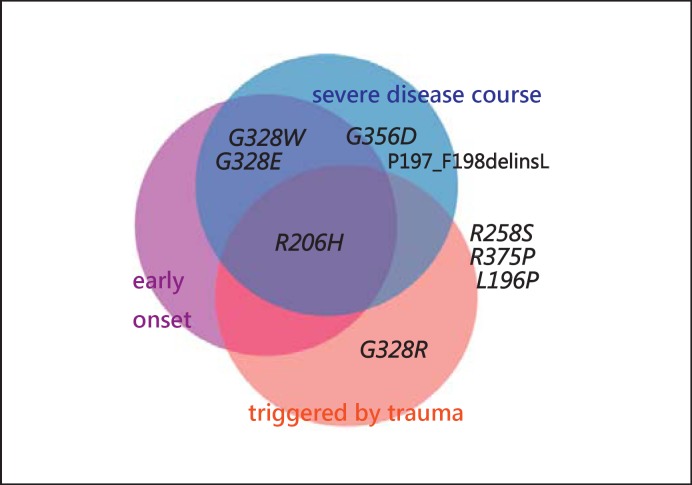
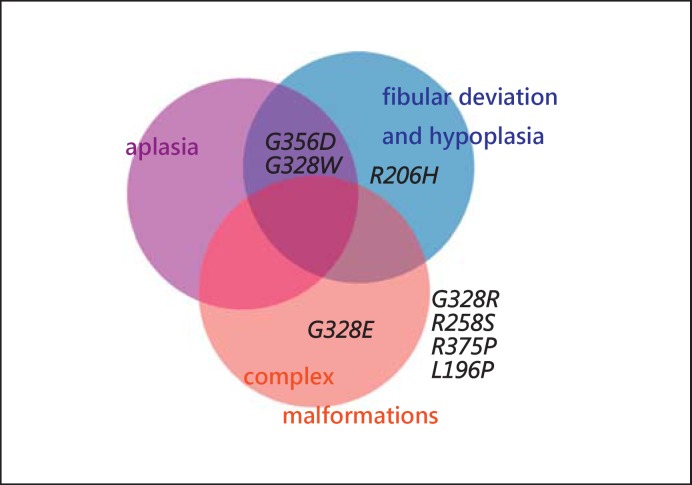
Fibrodysplasia ossificans progressiva (FOP, MIM 135100) is a rare autosomal dominant genetic disorder and the most disabling condition of heterotopic (extraskeletal) ossification in humans. Mutations in the ACVR1 gene (MIM 102576) were identified as a genetic cause of FOP [Shore et al., 2006]. Most patients with FOP have the same recurrent single nucleotide change c.617G>A, p.R206H in the ACVR1 gene. Furthermore, 11 other mutations in the ACVR1 gene have been described as a cause of FOP. Here, we review phenotypic and molecular findings of 130 cases of FOP reported in the literature from 1982 to April 2014 and discuss possible genotype-phenotype correlations in FOP patients.
Keywords: ACVR1; FOP; Great toe malformations; Heterotopic ossifications; Progressive immobility.
Figures
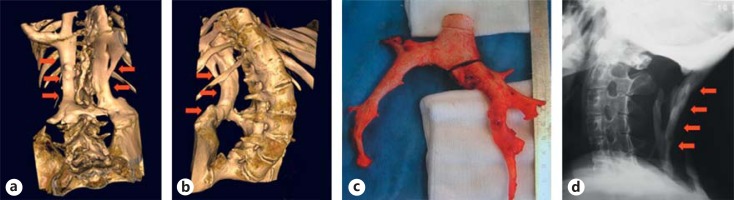
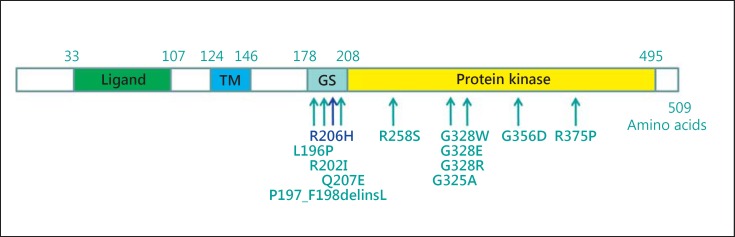
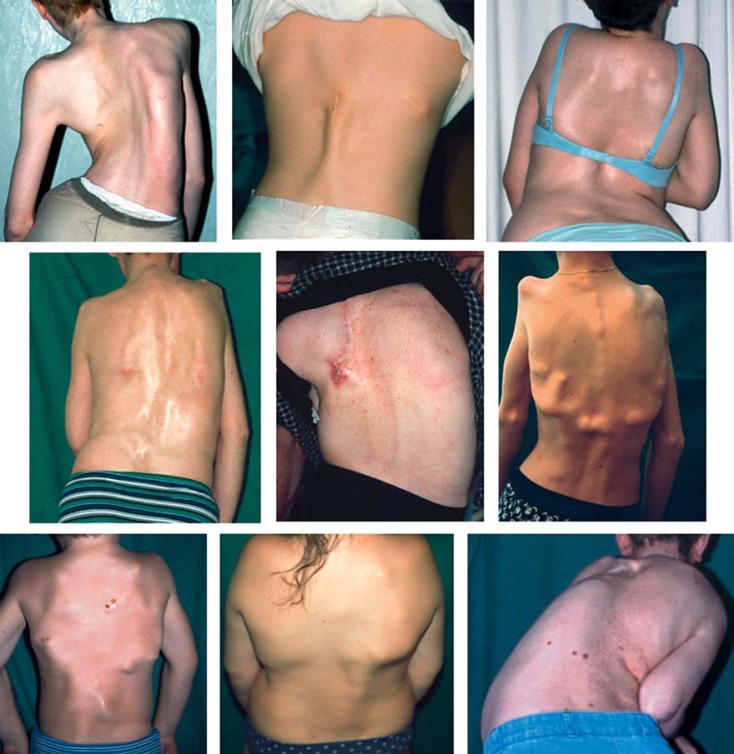
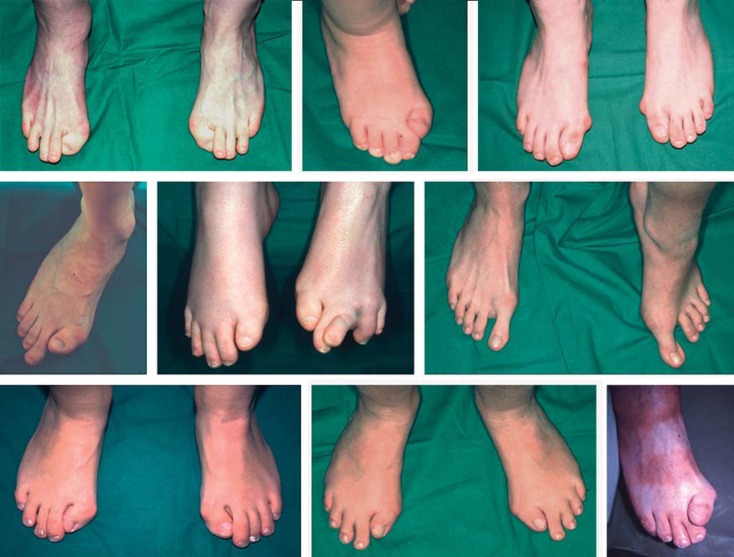
References
-
- Bauer KH, Bode W. Erbpathologie der Stützgewebe beim Menschen. Erbbiologie und Erbpathologie körperlicher Zustände und Funktionen. 1940;1:105.
-
- Carvalho DR, Navarro MM, Martins BJ, Coelho KE, Mello WD, et al. Mutational screening of ACVR1 gene in Brazilian fibrodysplasia ossificans progressiva patients. Clin Genet. 2010;77:171–176. - PubMed
-
- Chen D, Zhao M, Mundy GR. Bone morphogenetic proteins. Growth Factors. 2004;22:233–241. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
