

Error estimates for the analysis of differential expression from RNA-seq count data
- PMID: 25337456
- PMCID: PMC4179614
- DOI: 10.7717/peerj.576
Error estimates for the analysis of differential expression from RNA-seq count data
Abstract
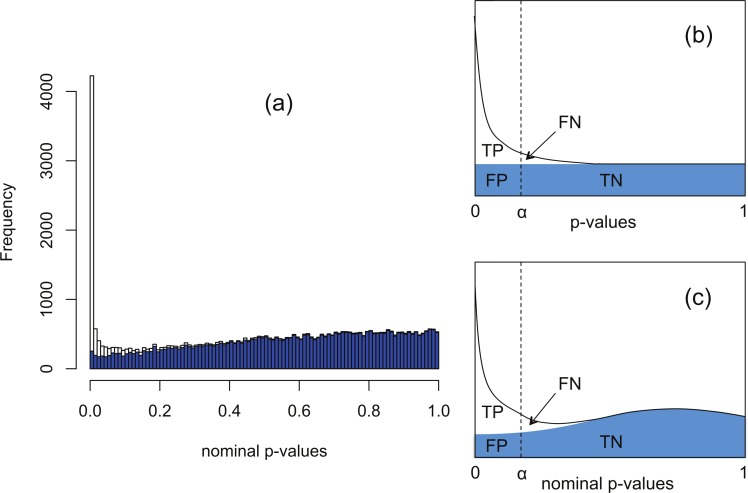
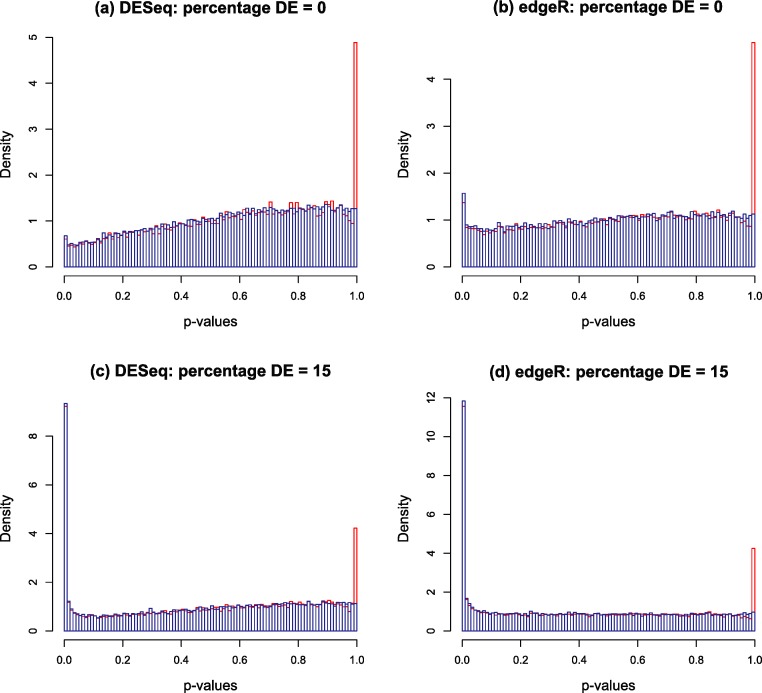
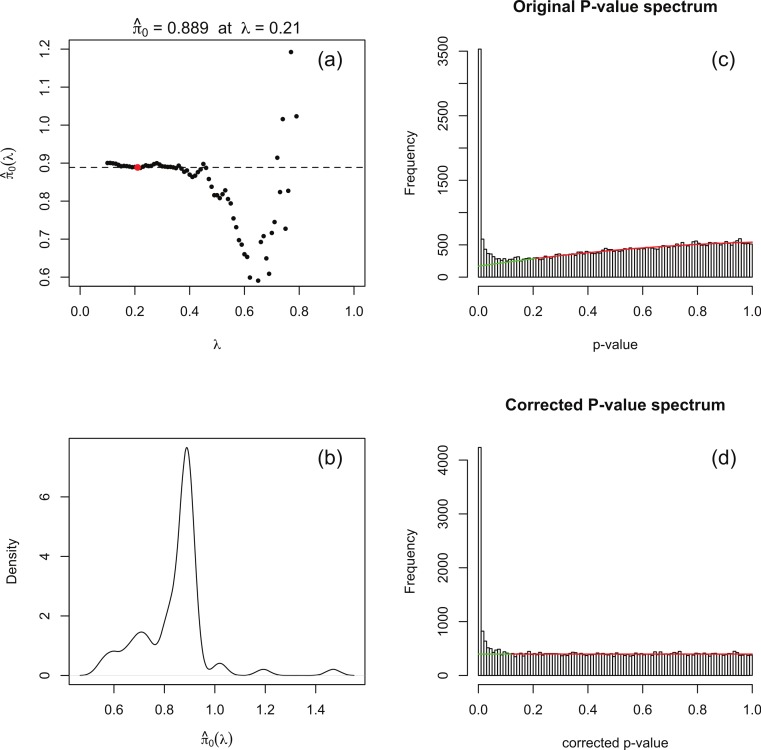
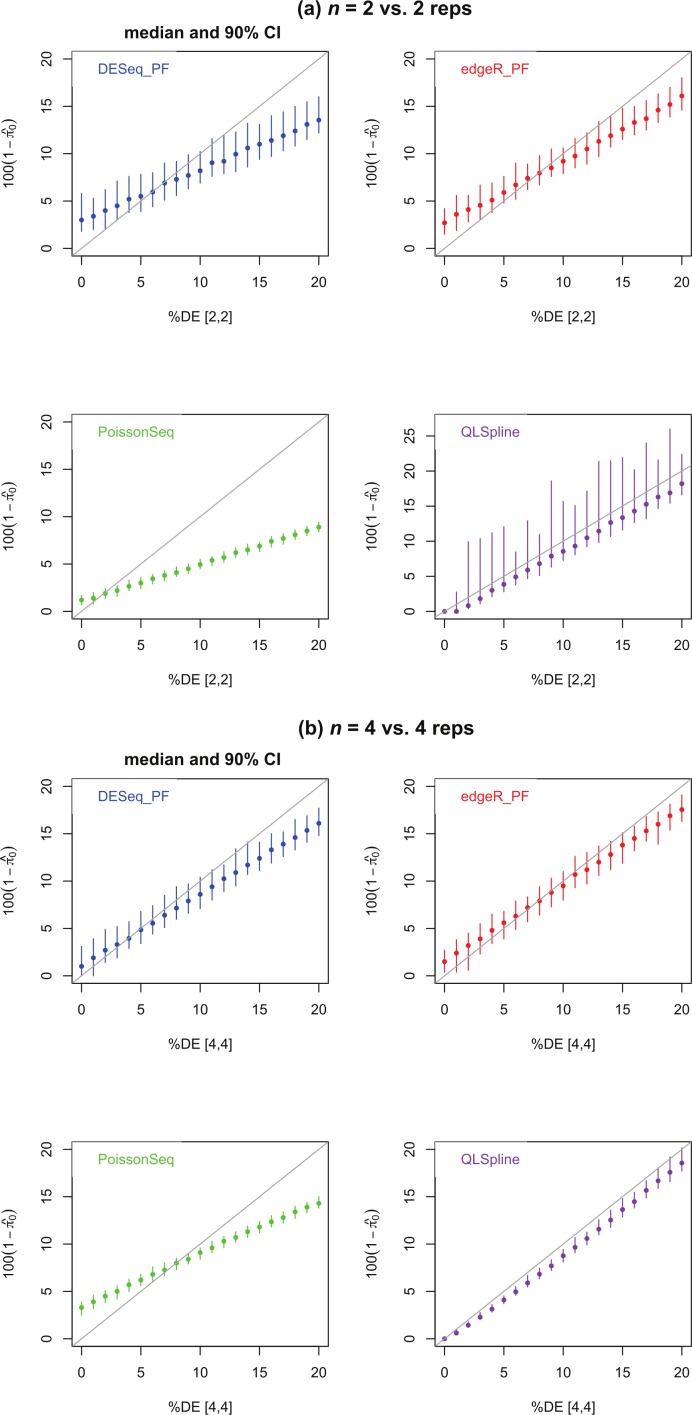
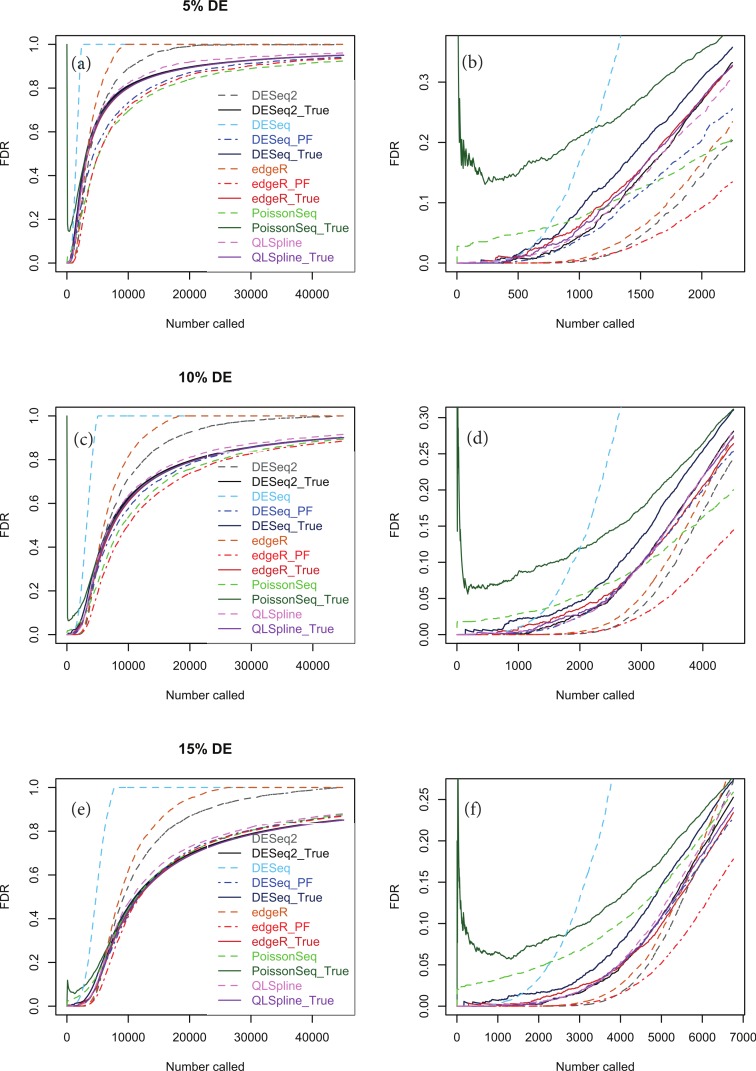
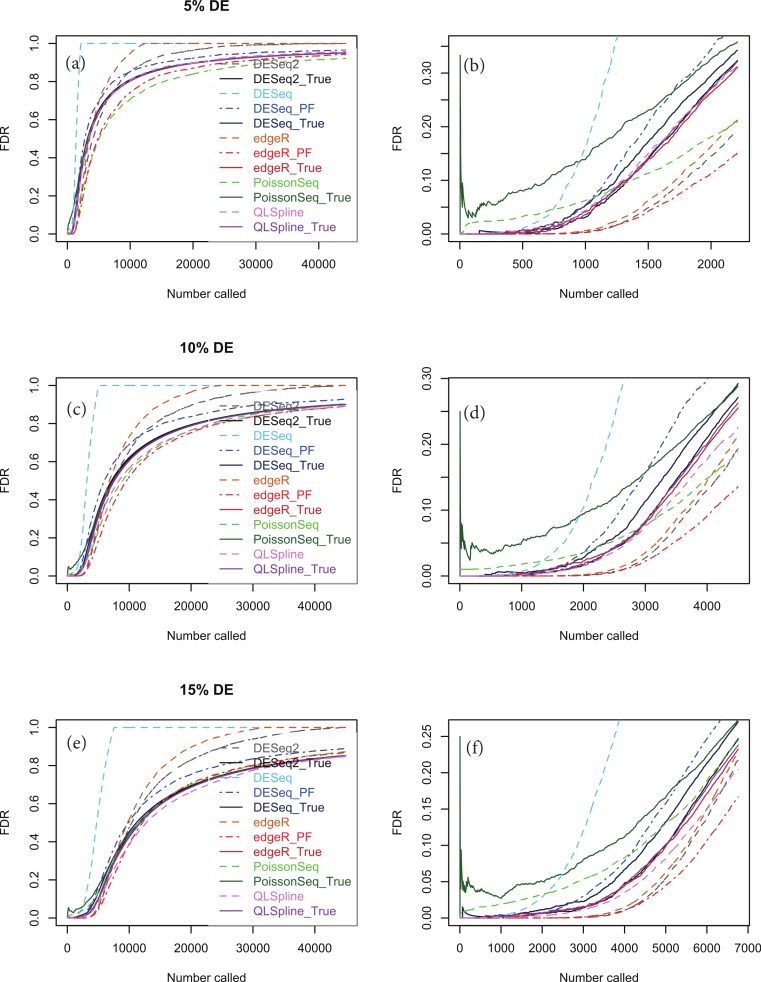
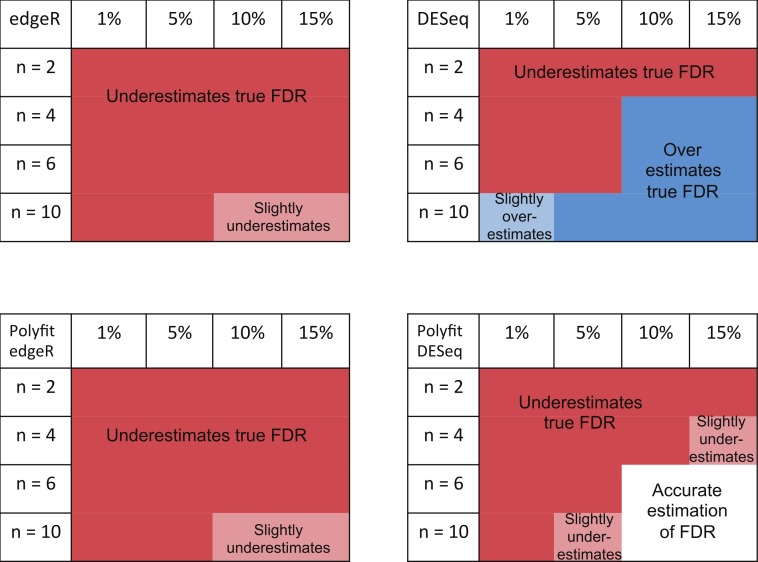
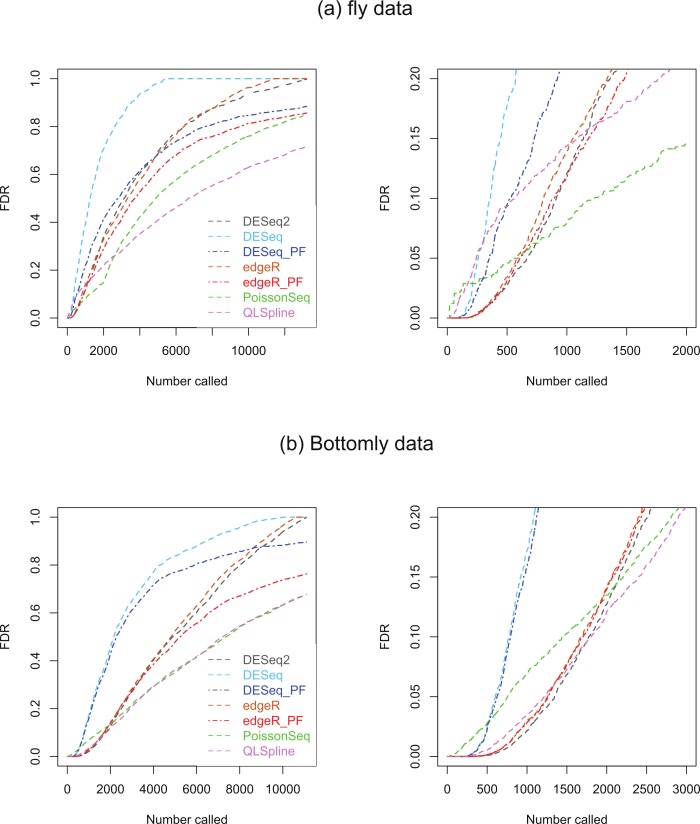
Background. A number of algorithms exist for analysing RNA-sequencing data to infer profiles of differential gene expression. Problems inherent in building algorithms around statistical models of over dispersed count data are formidable and frequently lead to non-uniform p-value distributions for null-hypothesis data and to inaccurate estimates of false discovery rates (FDRs). This can lead to an inaccurate measure of significance and loss of power to detect differential expression. Results. We use synthetic and real biological data to assess the ability of several available R packages to accurately estimate FDRs. The packages surveyed are based on statistical models of overdispersed Poisson data and include edgeR, DESeq, DESeq2, PoissonSeq and QuasiSeq. Also tested is an add-on package to edgeR and DESeq which we introduce called Polyfit. Polyfit aims to address the problem of a non-uniform null p-value distribution for two-class datasets by adapting the Storey-Tibshirani procedure. Conclusions. We find the best performing package in the sense that it achieves a low FDR which is accurately estimated over the full range of p-values, albeit with a very slow run time, is the QLSpline implementation of QuasiSeq. This finding holds provided the number of biological replicates in each condition is at least 4. The next best performing packages are edgeR and DESeq2. When the number of biological replicates is sufficiently high, and within a range accessible to multiplexed experimental designs, the Polyfit extension improves the performance DESeq (for approximately 6 or more replicates per condition), making its performance comparable with that of edgeR and DESeq2 in our tests with synthetic data.
Keywords: Differential expression analysis; False discovery rates; RNA-seq.
Figures
References
-
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Proceedings of the Royal Statistical Society Series B. 1995;57:289–300.
-
- Dillies M-A, Rau A, Aubert J, Hennequet-Antier C, Jeanmougin M, Servant N, Keime C, Marot G, Castel D, Estelle J, Guernec G, Jagla B, Jouneau L, Laloë D, Le Gall C, Schaëffer B, Le Crom S, Guedj M, Jaffrézic F. A comprehensive evaluation of normalization methods for illumina high-throughput RNA sequencing data analysis. Briefings in Bioinformatics. 2013;14:671–683. doi: 10.1093/bib/bbs046. - DOI - PubMed
-
- Dunne A, Pawitan Y, Doody L. Two-sided P-values from discrete asymmetric distributions based on uniformly most powerful unbiased tests. The Statistician. 1996;45(4):397–405. doi: 10.2307/2988542. - DOI
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
