

The new biology of estrogen-induced apoptosis applied to treat and prevent breast cancer
- PMID: 25339261
- PMCID: PMC4494663
- DOI: 10.1530/ERC-14-0448
The new biology of estrogen-induced apoptosis applied to treat and prevent breast cancer
Abstract
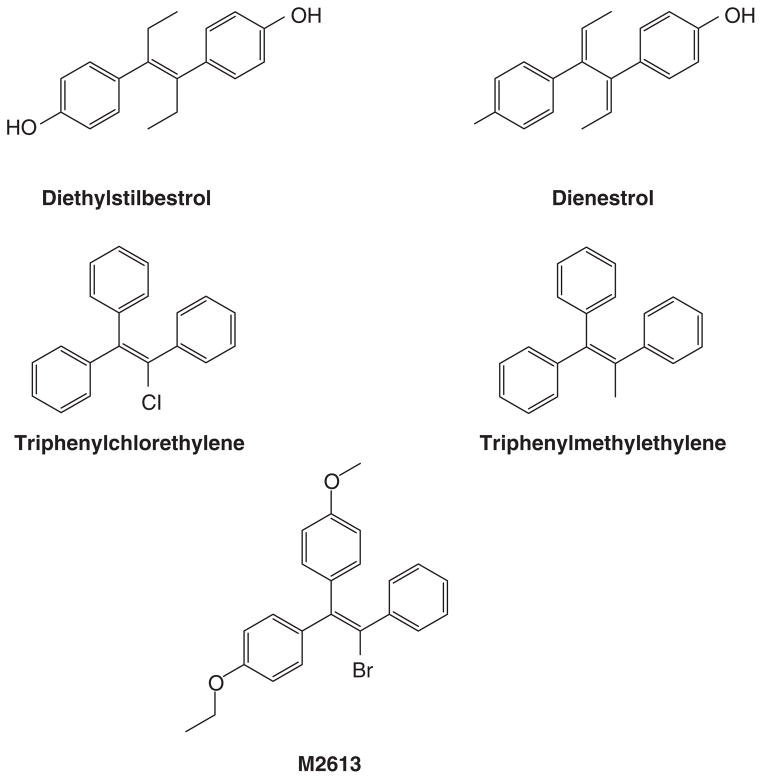
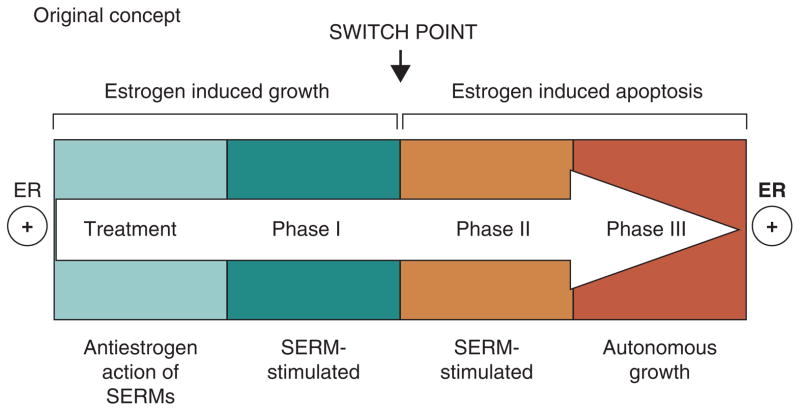
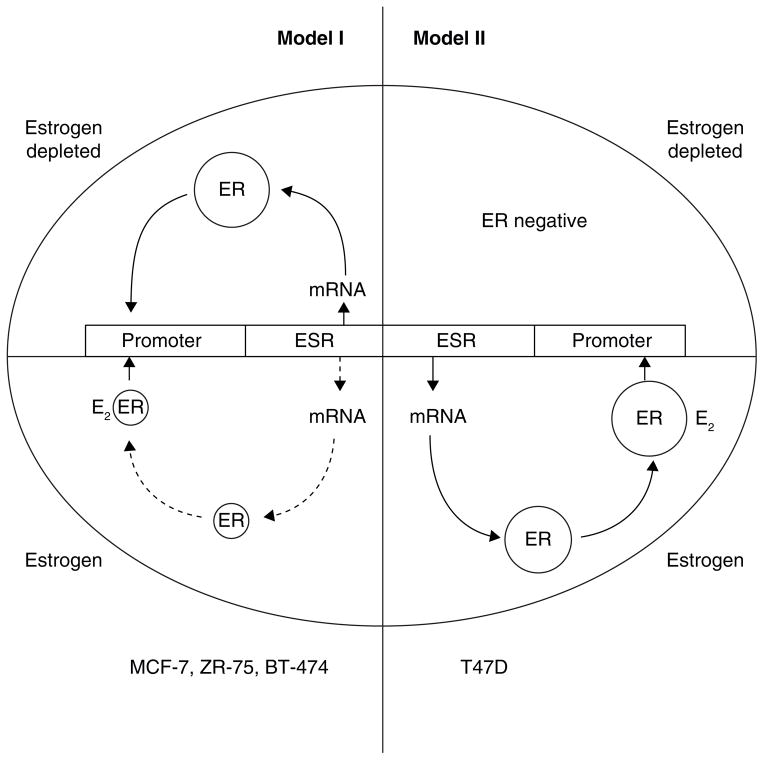
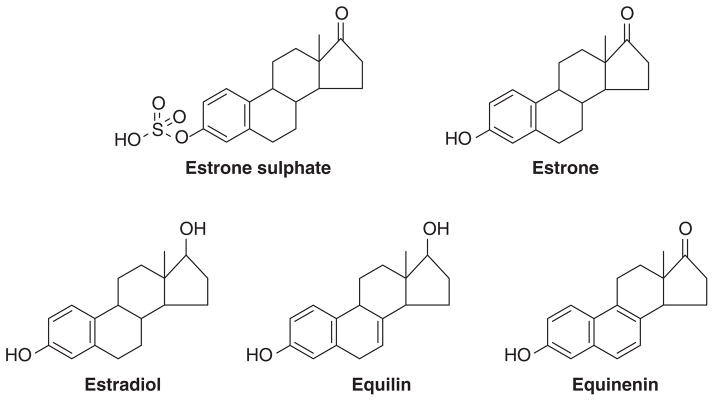
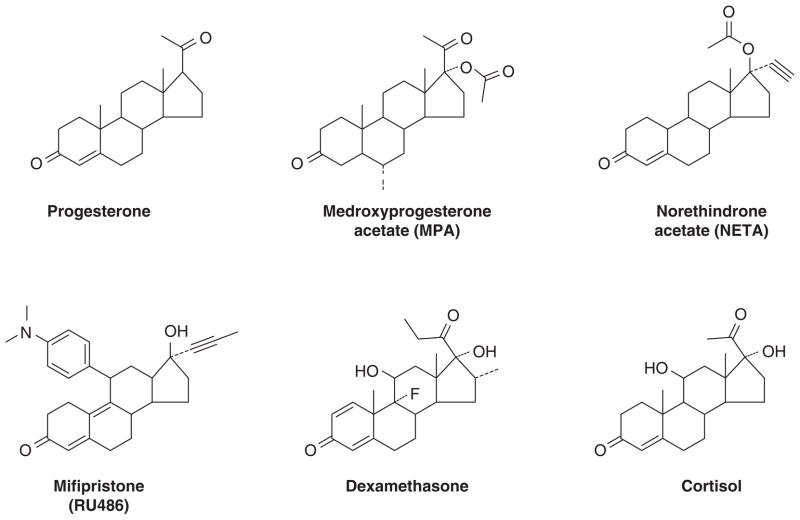
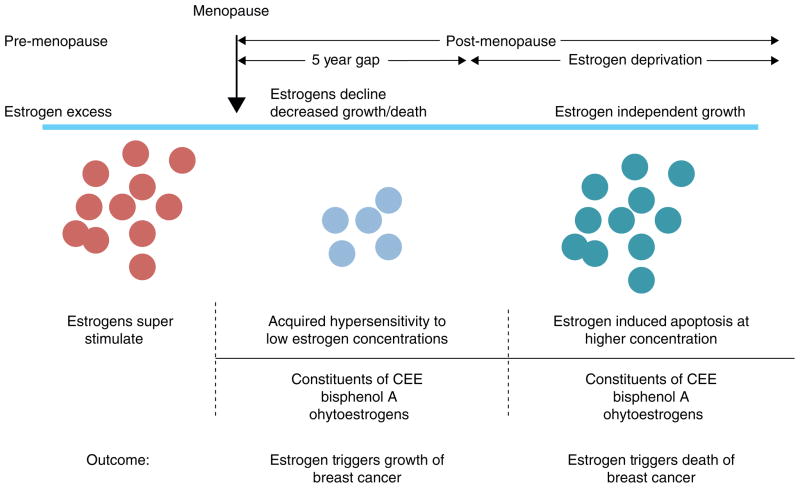
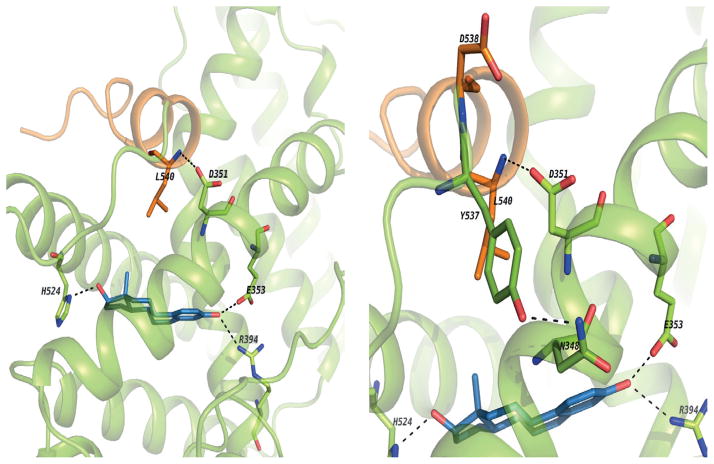
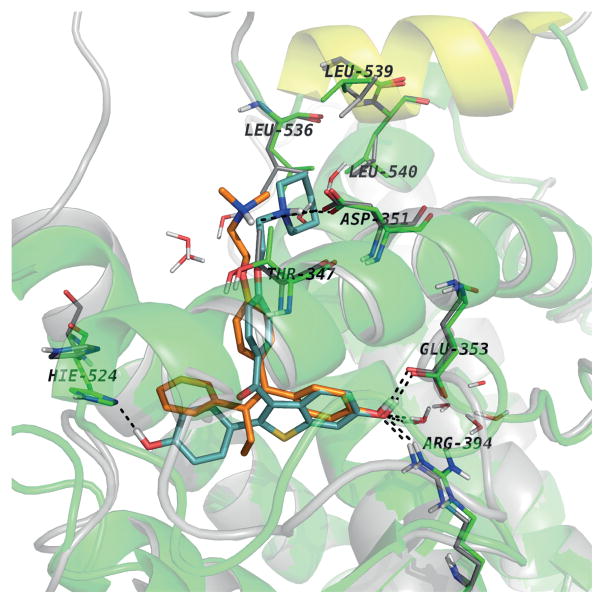
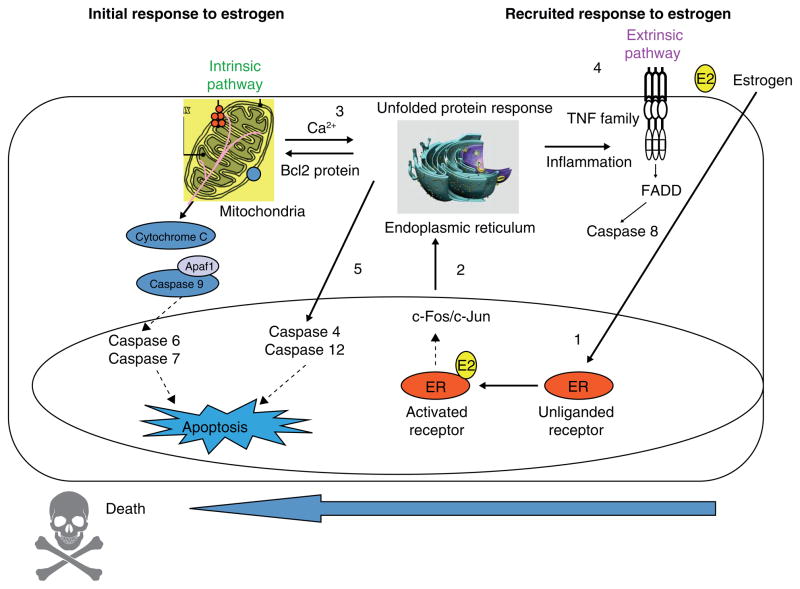
The successful use of high-dose synthetic estrogens to treat postmenopausal metastatic breast cancer is the first effective 'chemical therapy' proven in clinical trial to treat any cancer. This review documents the clinical use of estrogen for breast cancer treatment or estrogen replacement therapy (ERT) in postmenopausal hysterectomized women, which can either result in breast cancer cell growth or breast cancer regression. This has remained a paradox since the 1950s until the discovery of the new biology of estrogen-induced apoptosis at the end of the 20th century. The key to triggering apoptosis with estrogen is the selection of breast cancer cell populations that are resistant to long-term estrogen deprivation. However, estrogen-independent growth occurs through trial and error. At the cellular level, estrogen-induced apoptosis is dependent upon the presence of the estrogen receptor (ER), which can be blocked by nonsteroidal or steroidal antiestrogens. The shape of an estrogenic ligand programs the conformation of the ER complex, which, in turn, can modulate estrogen-induced apoptosis: class I planar estrogens (e.g., estradiol) trigger apoptosis after 24 h, whereas class II angular estrogens (e.g., bisphenol triphenylethylene) delay the process until after 72 h. This contrasts with paclitaxel, which causes G2 blockade with immediate apoptosis. The process is complete within 24 h. Estrogen-induced apoptosis is modulated by glucocorticoids and cSrc inhibitors, but the target mechanism for estrogen action is genomic and not through a nongenomic pathway. The process is stepwise through the creation of endoplasmic reticulum stress and inflammatory responses, which then initiate an unfolded protein response. This, in turn, initiates apoptosis through the intrinsic pathway (mitochondrial) with the subsequent recruitment of the extrinsic pathway (death receptor) to complete the process. The symmetry of the clinical and laboratory studies now permits the creation of rules for the future clinical application of ERT or phytoestrogen supplements: a 5-year gap is necessary after menopause to permit the selection of estrogen-deprived breast cancer cell populations to cause them to become vulnerable to apoptotic cell death. Earlier treatment with estrogen around menopause encourages growth of ER-positive tumor cells, as the cells are still dependent on estrogen to maintain replication within the expanding population. An awareness of the evidence that the molecular events associated with estrogen-induced apoptosis can be orchestrated in the laboratory in estrogen-deprived breast cancers now supports the clinical findings regarding the treatment of metastatic breast cancer following estrogen deprivation, decreases in mortality following long-term antihormonal adjuvant therapy, and the results of treatment with ERT and ERT plus progestin in the Women's Health Initiative for women over the age of 60. Principles have emerged for understanding and applying physiological estrogen therapy appropriately by targeting the correct patient populations.
Keywords: acquired resistance; aromatase inhibitors; raloxifene; selective estrogen receptor modulators; tamoxifen.
© 2015 Society for Endocrinology.
Figures

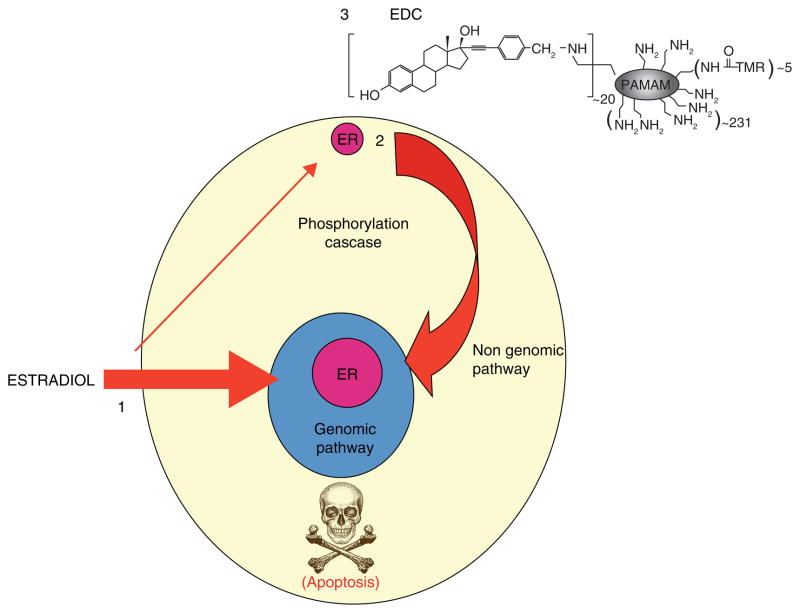
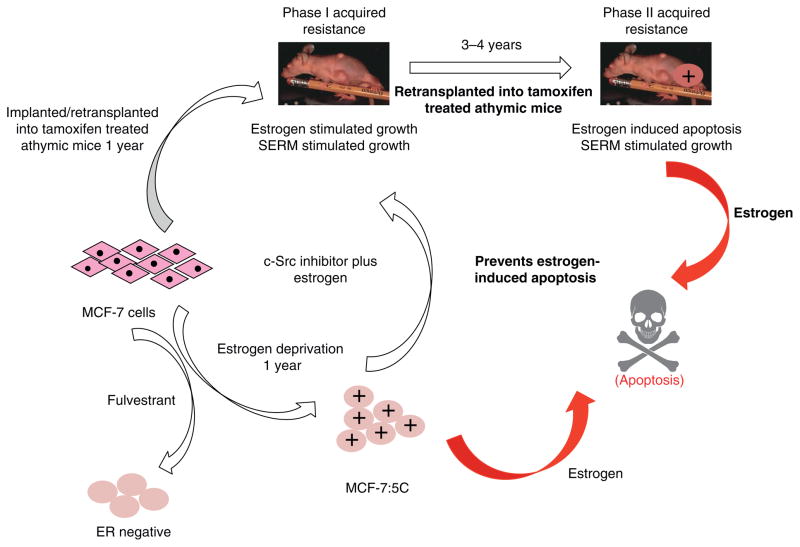
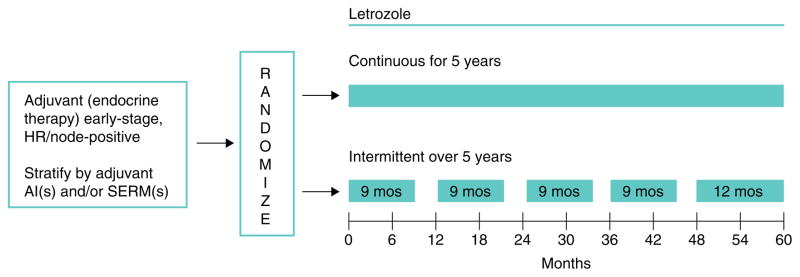
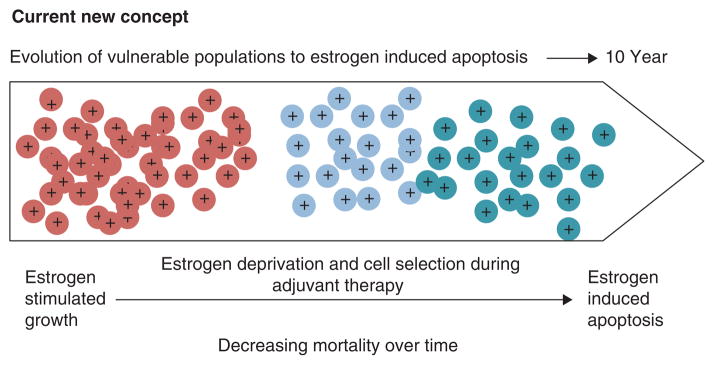
References
-
- Ingle JN, Ahmann DL, Green SJ, et al. Randomized clinical trial of diethylstilbestrol versus tamoxifen in postmenopausal women with advanced breast cancer. N Engl J Med. 1981;304(1):16–21. - PubMed
-
- Kennedy BJ. Hormone therapy for advanced breast cancer. Cancer. 1965;18(12):1551–7. - PubMed
-
- Wolf DM, Jordan VC. A laboratory model to explain the survival advantage observed in patients taking adjuvant tamoxifen therapy. Recent Results Cancer Res. 1993;127:23–33. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
