

Regulation of presynaptic Ca2+, synaptic plasticity and contextual fear conditioning by a N-terminal β-amyloid fragment
- PMID: 25339735
- PMCID: PMC4205547
- DOI: 10.1523/JNEUROSCI.0326-14.2014
Regulation of presynaptic Ca2+, synaptic plasticity and contextual fear conditioning by a N-terminal β-amyloid fragment
Abstract
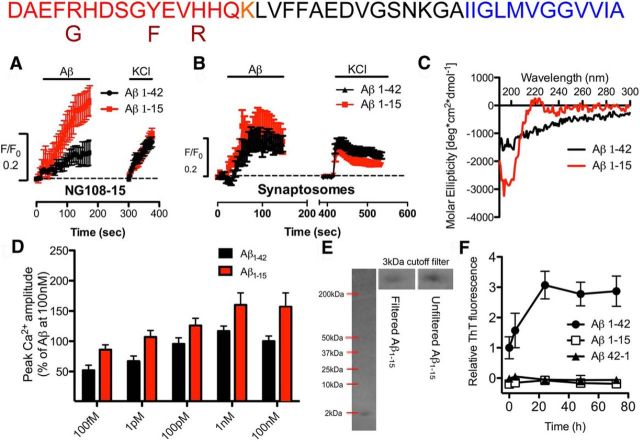
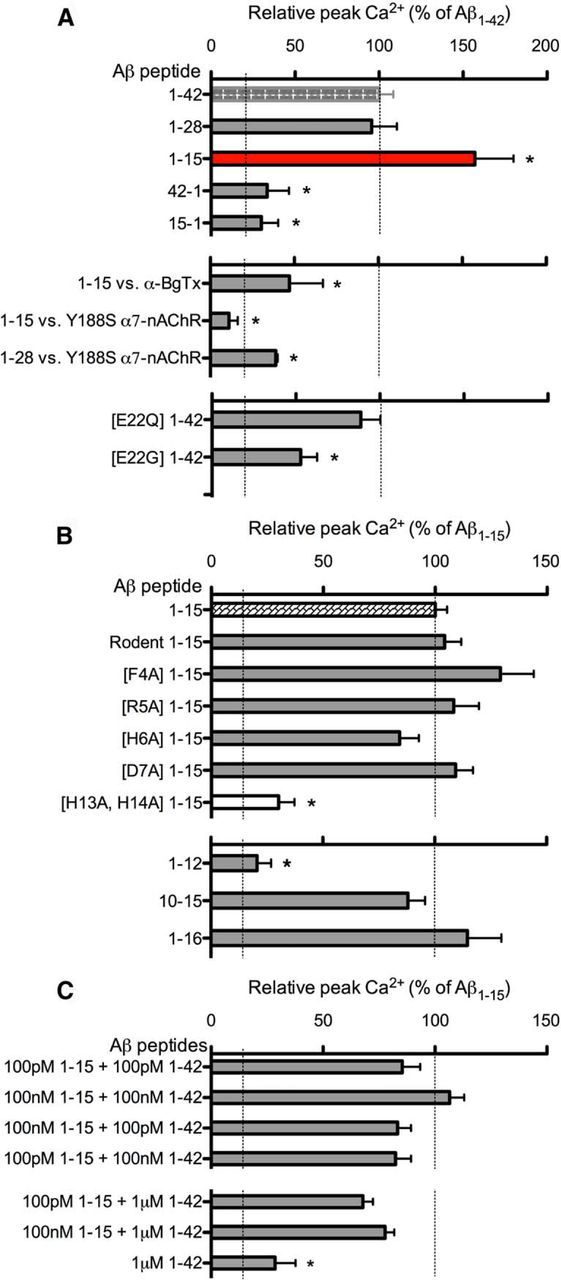
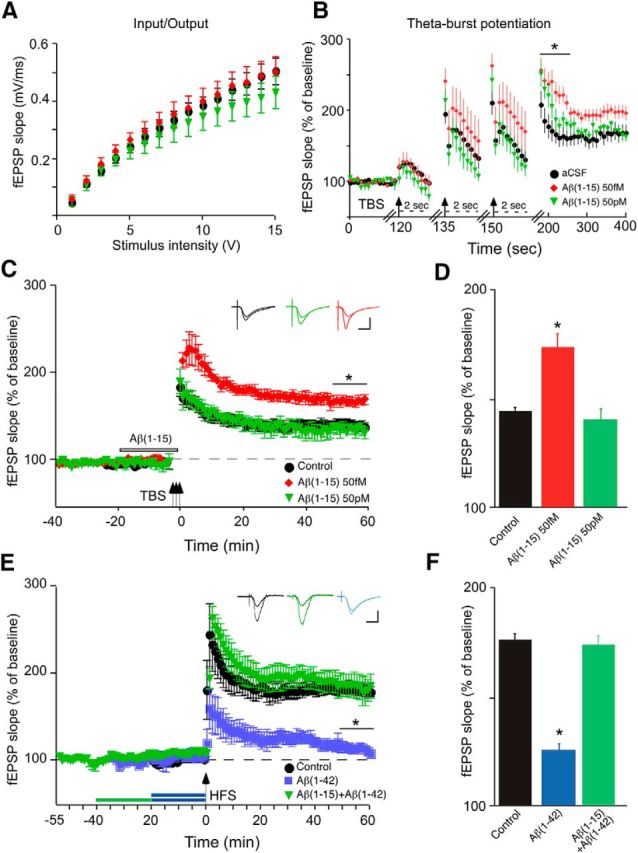
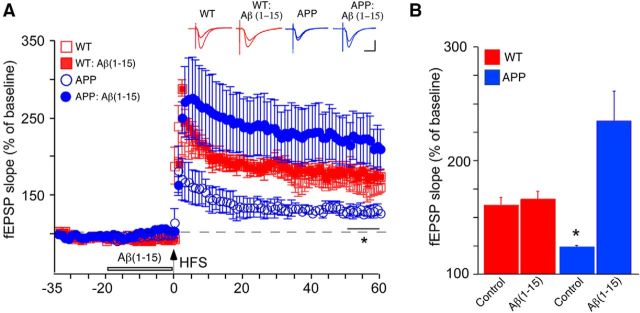
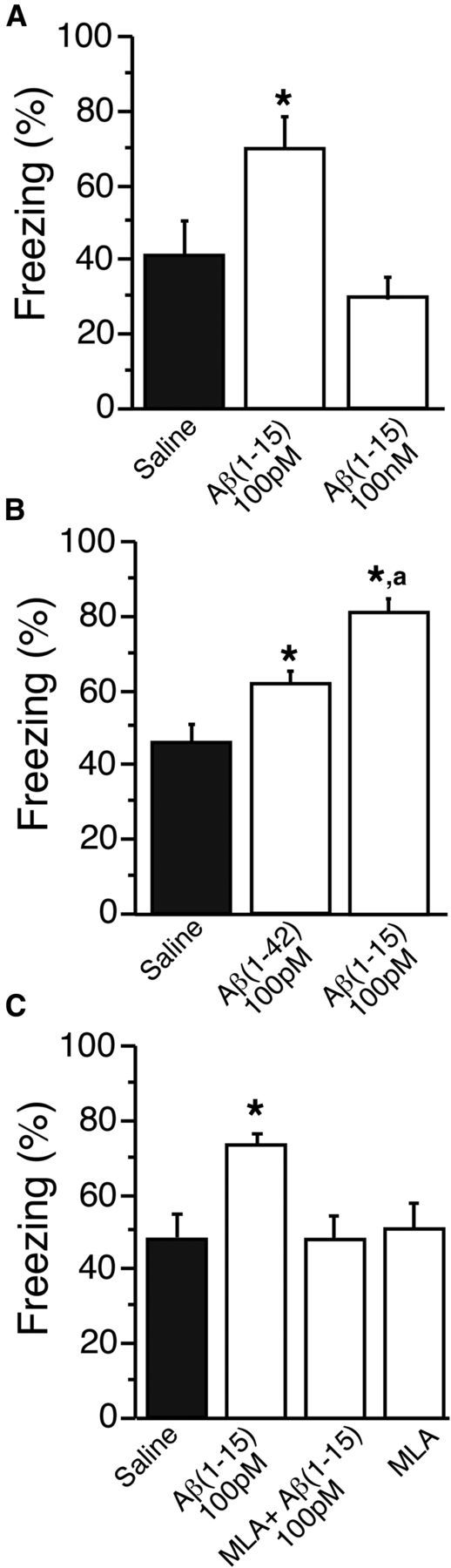
Soluble β-amyloid has been shown to regulate presynaptic Ca(2+) and synaptic plasticity. In particular, picomolar β-amyloid was found to have an agonist-like action on presynaptic nicotinic receptors and to augment long-term potentiation (LTP) in a manner dependent upon nicotinic receptors. Here, we report that a functional N-terminal domain exists within β-amyloid for its agonist-like activity. This sequence corresponds to a N-terminal fragment generated by the combined action of α- and β-secretases, and resident carboxypeptidase. The N-terminal β-amyloid fragment is present in the brains and CSF of healthy adults as well as in Alzheimer's patients. Unlike full-length β-amyloid, the N-terminal β-amyloid fragment is monomeric and nontoxic. In Ca(2+) imaging studies using a model reconstituted rodent neuroblastoma cell line and isolated mouse nerve terminals, the N-terminal β-amyloid fragment proved to be highly potent and more effective than full-length β-amyloid in its agonist-like action on nicotinic receptors. In addition, the N-terminal β-amyloid fragment augmented theta burst-induced post-tetanic potentiation and LTP in mouse hippocampal slices. The N-terminal fragment also rescued LTP inhibited by elevated levels of full-length β-amyloid. Contextual fear conditioning was also strongly augmented following bilateral injection of N-terminal β-amyloid fragment into the dorsal hippocampi of intact mice. The fragment-induced augmentation of fear conditioning was attenuated by coadministration of nicotinic antagonist. The activity of the N-terminal β-amyloid fragment appears to reside largely in a sequence surrounding a putative metal binding site, YEVHHQ. These findings suggest that the N-terminal β-amyloid fragment may serve as a potent and effective endogenous neuromodulator.
Keywords: Ca regulation; fear memory; neuromodulation; synaptic plasticity; β-amyloid.
Copyright © 2014 the authors 0270-6474/14/3414210-09$15.00/0.
Figures
References
-
- Chin JH, Ma L, MacTavish D, Jhamandas JH. Amyloid β protein modulates glutamate-mediated neurotransmission in the rat basal forebrain: involvement of presynaptic neuronal nicotinic acetylcholine and metabotropic glutamate receptors. J Neurosci. 2007;27:9262–9269. doi: 10.1523/JNEUROSCI.1843-07.2007. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous