

Inhibiting ACAT1/SOAT1 in microglia stimulates autophagy-mediated lysosomal proteolysis and increases Aβ1-42 clearance
- PMID: 25339759
- PMCID: PMC4205563
- DOI: 10.1523/JNEUROSCI.2567-14.2014
Inhibiting ACAT1/SOAT1 in microglia stimulates autophagy-mediated lysosomal proteolysis and increases Aβ1-42 clearance
Abstract
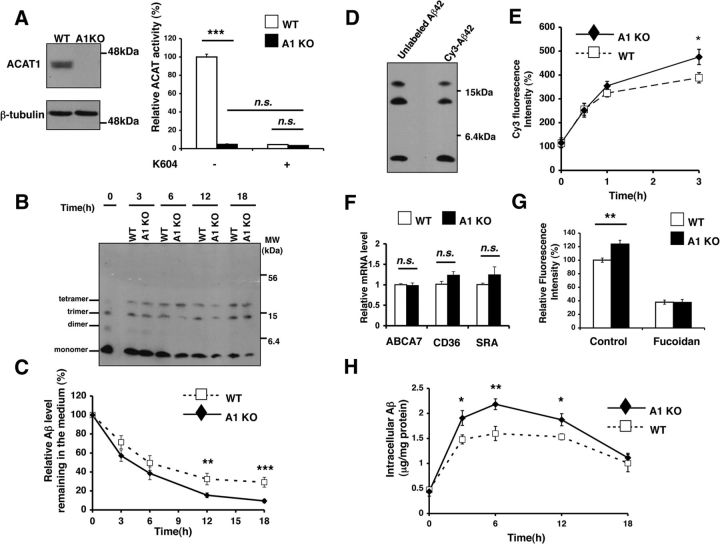
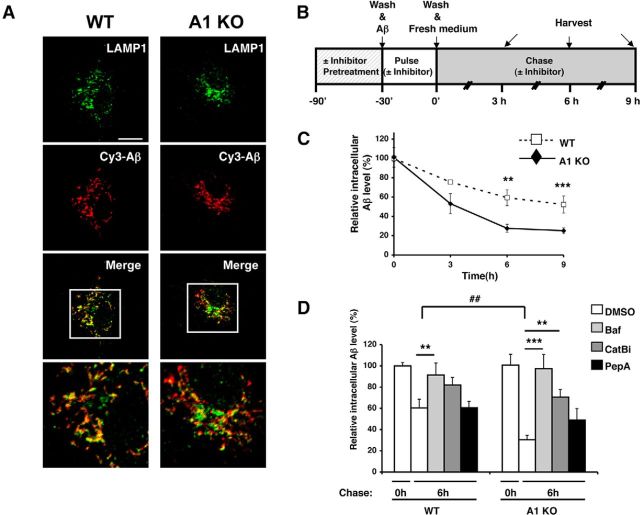
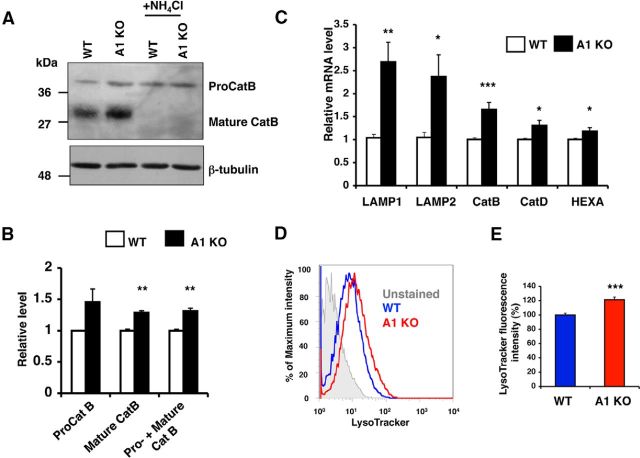
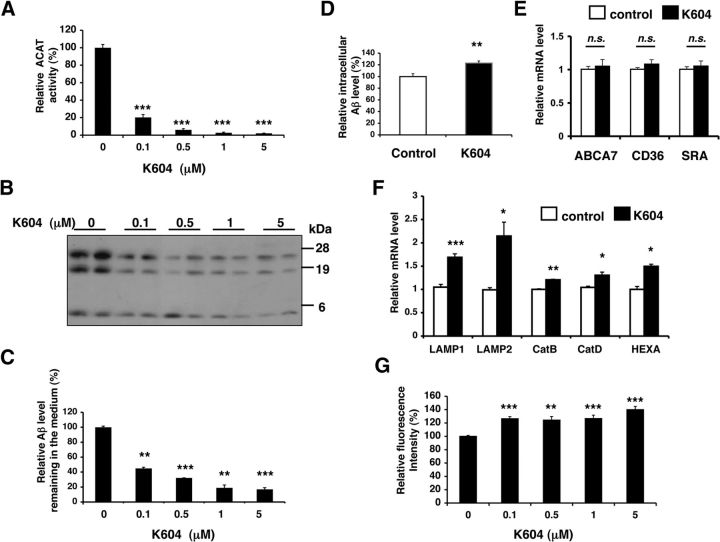
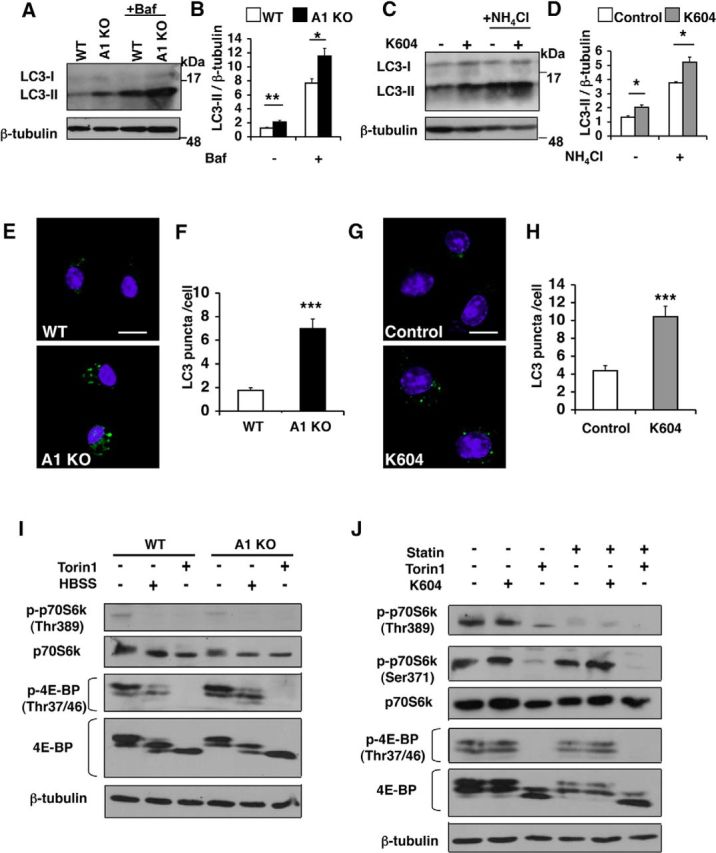
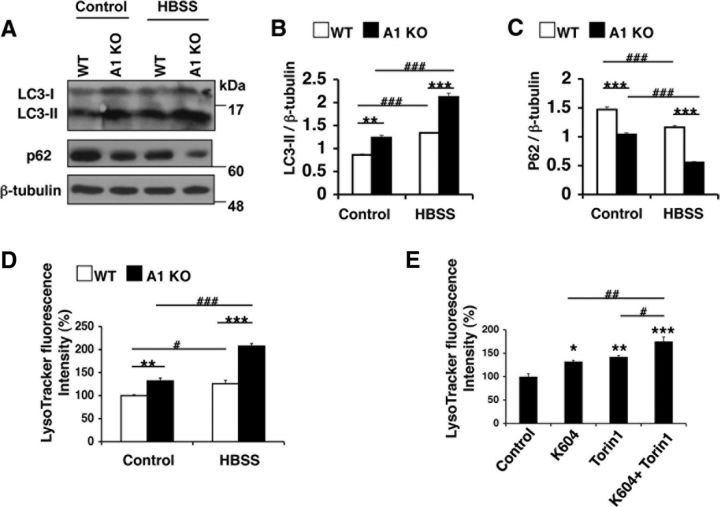
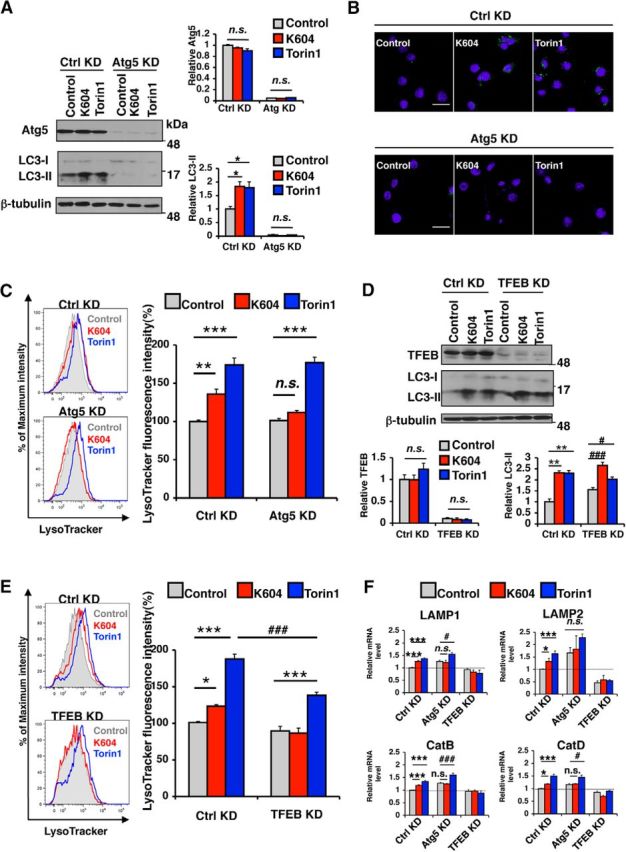
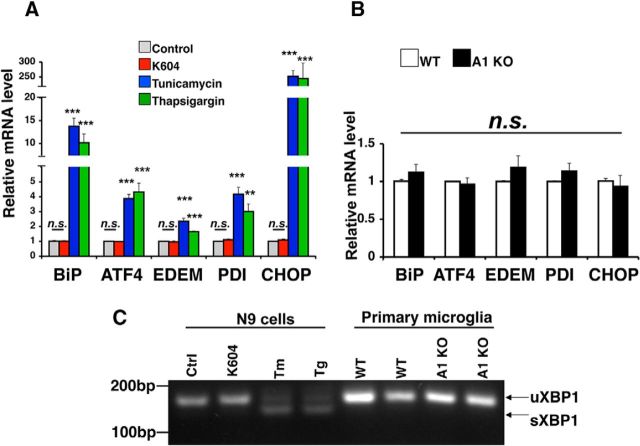
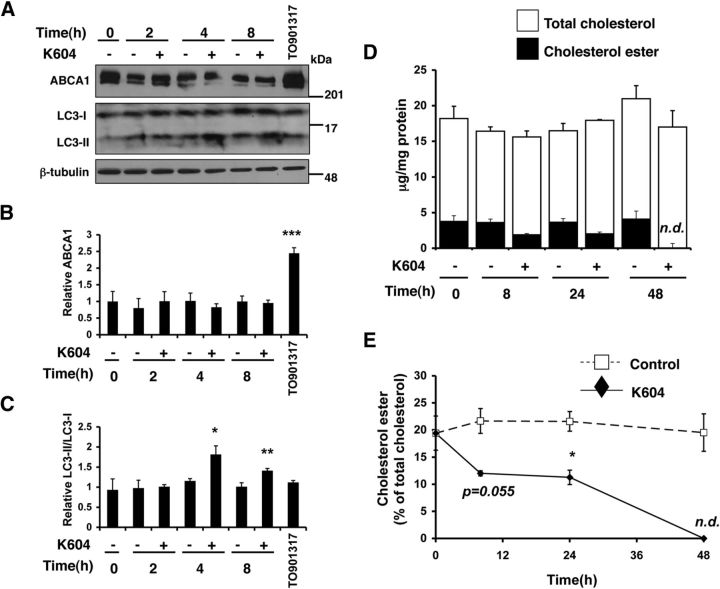
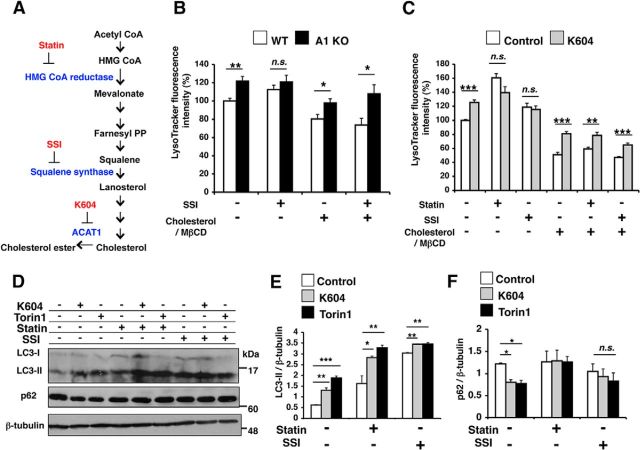
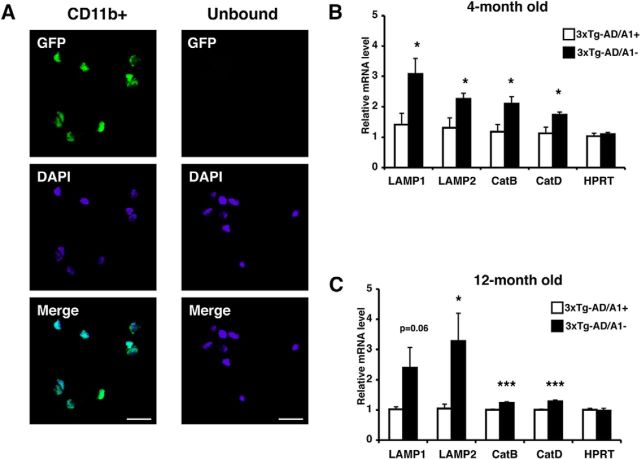
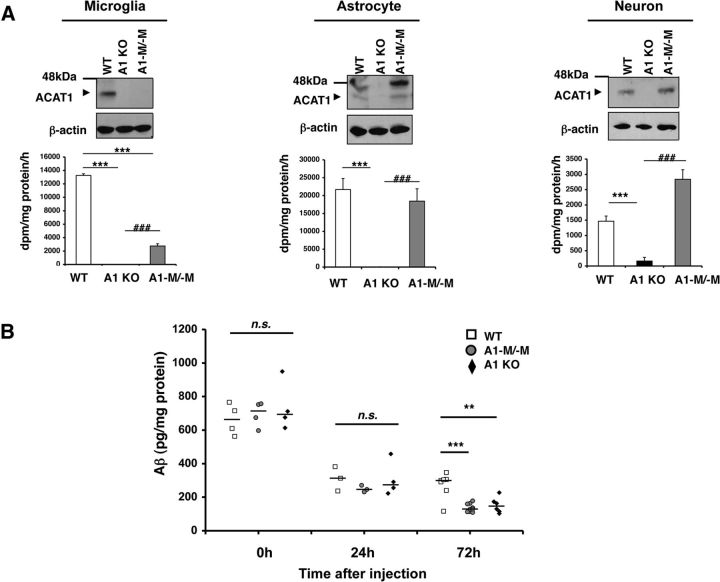
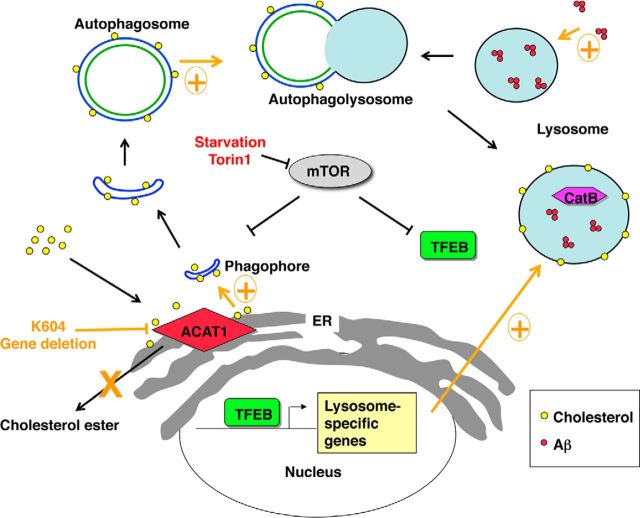
Acyl-CoA:cholesterol acyltransferase 1 (ACAT1) is a resident endoplasmic reticulum enzyme that prevents the buildup of cholesterol in membranes by converting it to cholesterol esters. Blocking ACAT1 pharmacologically or by Acat1 gene knock-out (KO) decreases amyloidopathy in mouse models for Alzheimer's disease. However, the beneficial actions of ACAT1 blockage to treat Alzheimer's disease remained not well understood. Microglia play essential roles in the proteolytic clearance of amyloid β (Aβ) peptides. Here we show that Acat1 gene KO in mouse increases phagocytic uptake of oligomeric Aβ1-42 and stimulates lysosomal Aβ1-42 degradation in cultured microglia and in vivo. Additional results show that Acat1 gene KO or a specific ACAT1 inhibitor K604 stimulates autophagosome formation and transcription factor EB-mediated lysosomal proteolysis. Surprisingly, the effect of ACAT1 blockage does not alter mTOR signaling or endoplasmic reticulum stress response but can be modulated by agents that disrupt cholesterol biosynthesis. To our knowledge, our current study provides the first example that a small molecule (K604) can promote autophagy in an mTOR-independent manner to activate the coordinated lysosomal expression and regulation network. Autophagy is needed to degrade misfolded proteins/peptides. Our results implicate that blocking ACAT1 may provide a new way to benefit multiple neurodegenerative diseases.
Keywords: ACAT; Alzheimer's disease; autophagy; cholesterol; microglia.
Copyright © 2014 the authors 0270-6474/14/3414484-18$15.00/0.
Figures
References
-
- Area-Gomez E, Del Carmen Lara Castillo M, Tambini MD, Guardia-Laguarta C, de Groof AJ, Madra M, Ikenouchi J, Umeda M, Bird TD, Sturley SL, Schon EA. Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. EMBO J. 2012;31:4106–4123. doi: 10.1038/emboj.2012.202. - DOI - PMC - PubMed
-
- Balducci C, Beeg M, Stravalaci M, Bastone A, Sclip A, Biasini E, Tapella L, Colombo L, Manzoni C, Borsello T, Chiesa R, Gobbi M, Salmona M, Forloni G. Synthetic amyloid-β oligomers impair long-term memory independently of cellular prion protein. Proc Natl Acad Sci U S A. 2010;107:2295–2300. doi: 10.1073/pnas.0911829107. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous