

Glutathionylation of the L-type Ca2+ channel in oxidative stress-induced pathology of the heart
- PMID: 25340983
- PMCID: PMC4227269
- DOI: 10.3390/ijms151019203
Glutathionylation of the L-type Ca2+ channel in oxidative stress-induced pathology of the heart
Abstract
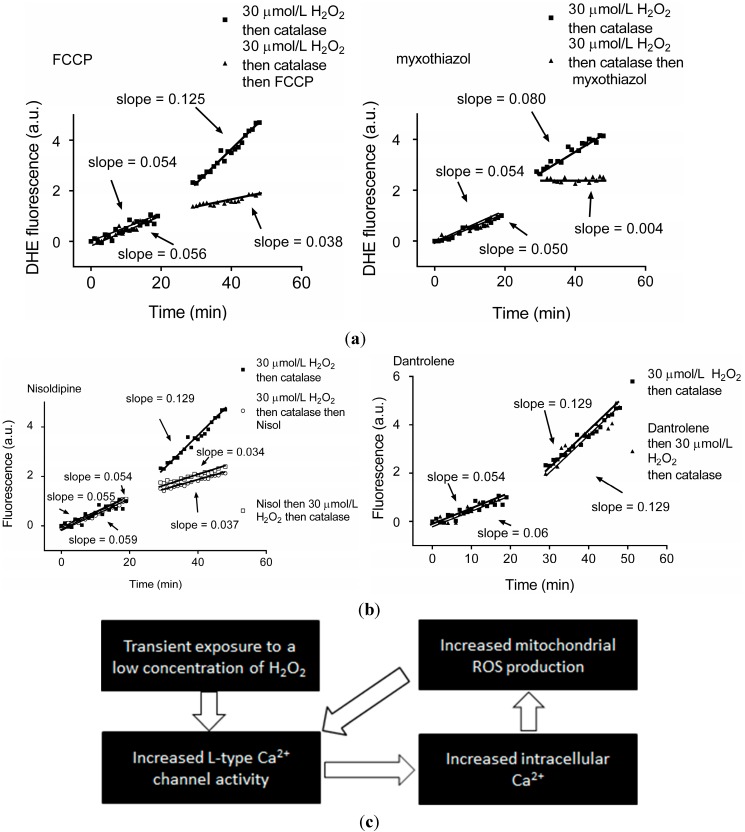
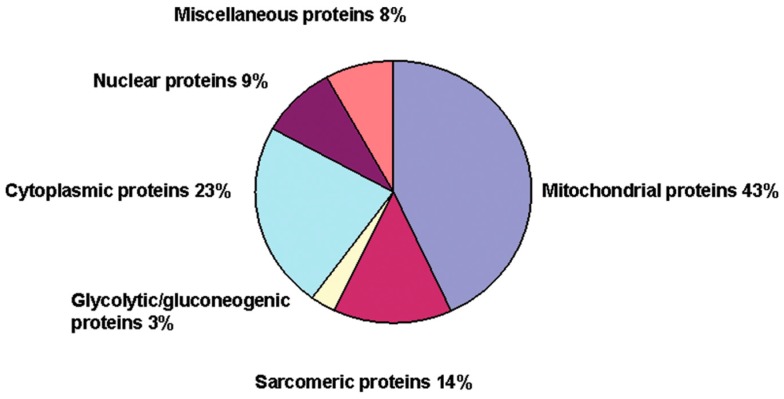
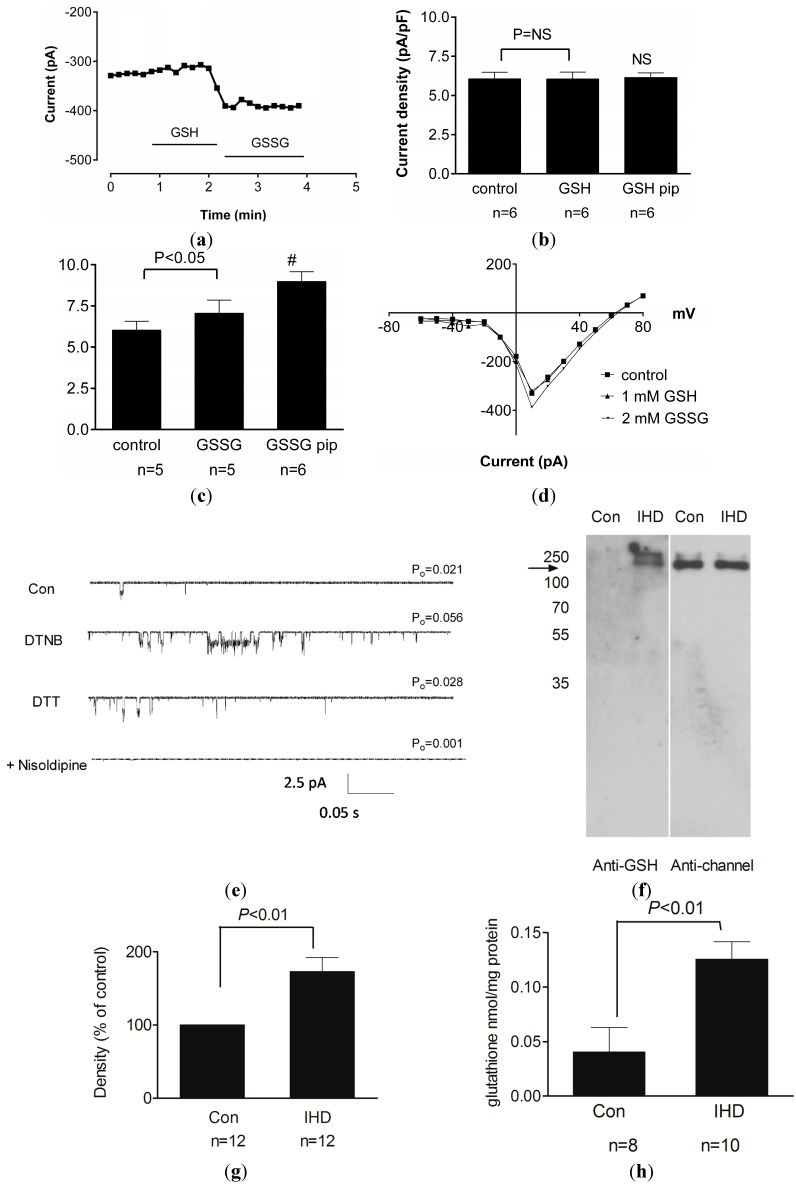
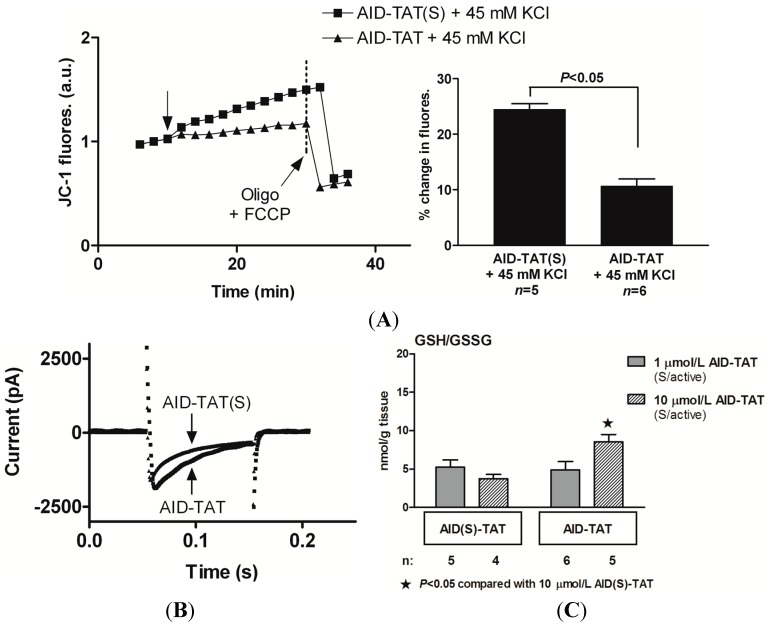
There is mounting evidence to suggest that protein glutathionylation is a key process contributing to the development of pathology. Glutathionylation occurs as a result of posttranslational modification of a protein and involves the addition of a glutathione moiety at cysteine residues. Such modification can occur on a number of proteins, and exerts a variety of functional consequences. The L-type Ca2+ channel has been identified as a glutathionylation target that participates in the development of cardiac pathology. Ca2+ influx via the L-type Ca2+ channel increases production of mitochondrial reactive oxygen species (ROS) in cardiomyocytes during periods of oxidative stress. This induces a persistent increase in channel open probability, and the resulting constitutive increase in Ca2+ influx amplifies the cross-talk between the mitochondria and the channel. Novel strategies utilising targeted peptide delivery to uncouple mitochondrial ROS and Ca2+ flux via the L-type Ca2+ channel following ischemia-reperfusion have delivered promising results, and have proven capable of restoring appropriate mitochondrial function in myocytes and in vivo.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
