

Alzheimer's disease and prion protein
- PMID: 25343100
- PMCID: PMC4204584
- DOI: 10.5582/irdr.2013.v2.2.35
Alzheimer's disease and prion protein
Abstract
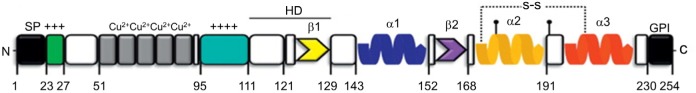
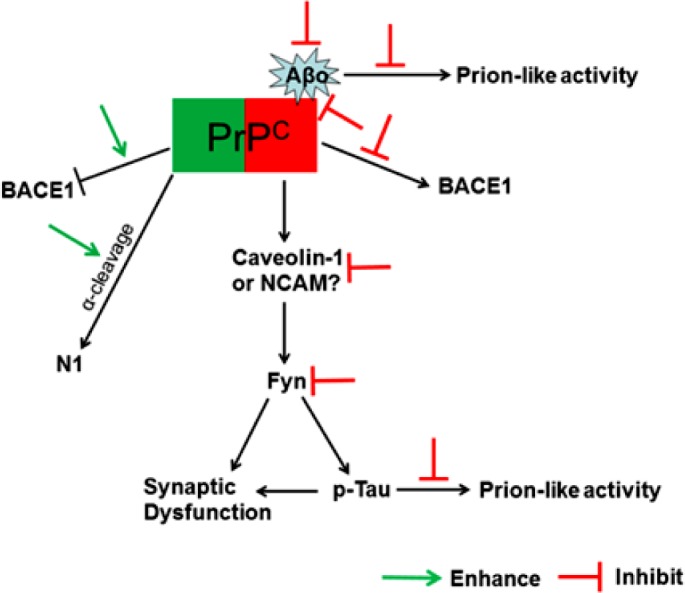
Alzheimer's disease (AD) is a devastating neurodegenerative disease with progressive loss of memory and cognitive function, pathologically hallmarked by aggregates of the amyloid-beta (Aβ) peptide and hyperphosphorylated tau in the brain. Aggregation of Aβ under the form of amyloid fibrils has long been considered central to the pathogenesis of AD. However, recent evidence has indicated that soluble Aβ oligomers, rather than insoluble fibrils, are the main neurotoxic species in AD. The cellular prion protein (PrP(C)) has newly been identified as a cell surface receptor for Aβ oligomers. PrP(C) is a cell surface glycoprotein that plays a key role in the propagation of prions, proteinaceous infectious agents that replicate by imposing their abnormal conformation to PrP(C) molecules. In AD, PrP(C) acts to transduce the neurotoxic signals arising from Aβ oligomers, leading to synaptic failure and cognitive impairment. Interestingly, accumulating evidence has also shown that aggregated Aβ or tau possesses prion-like activity, a property that would allow them to spread throughout the brain. In this article, we review recent findings regarding the function of PrP(C) and its role in AD, and discuss potential therapeutic implications of PrP(C)-based approaches in the treatment of AD.
Keywords: Fyn kinase; N1 fragment; PRNP gene; long-term potentiation (LTP); protein misfolding.
Figures
Similar articles
-
Soluble prion protein and its N-terminal fragment prevent impairment of synaptic plasticity by Aβ oligomers: Implications for novel therapeutic strategy in Alzheimer's disease.Neurobiol Dis. 2016 Jul;91:124-131. doi: 10.1016/j.nbd.2016.03.001. Epub 2016 Mar 3. Neurobiol Dis. 2016. PMID: 26949218 Free PMC article.
-
Prion protein prevents the inhibition of large-conductance calcium-activated potassium channel by Tau peptide K18 oligomers.Biochem Biophys Res Commun. 2024 Nov 19;734:150793. doi: 10.1016/j.bbrc.2024.150793. Epub 2024 Oct 8. Biochem Biophys Res Commun. 2024. PMID: 39378784
-
Exosomal cellular prion protein drives fibrillization of amyloid beta and counteracts amyloid beta-mediated neurotoxicity.J Neurochem. 2016 Apr;137(1):88-100. doi: 10.1111/jnc.13514. Epub 2016 Mar 2. J Neurochem. 2016. PMID: 26710111
-
Binding between Prion Protein and Aβ Oligomers Contributes to the Pathogenesis of Alzheimer's Disease.Virol Sin. 2019 Oct;34(5):475-488. doi: 10.1007/s12250-019-00124-1. Epub 2019 May 15. Virol Sin. 2019. PMID: 31093882 Free PMC article. Review.
-
Cellular Prion Protein as a Receptor of Toxic Amyloid-β42 Oligomers Is Important for Alzheimer's Disease.Front Cell Neurosci. 2019 Jul 30;13:339. doi: 10.3389/fncel.2019.00339. eCollection 2019. Front Cell Neurosci. 2019. PMID: 31417361 Free PMC article. Review.
Cited by
-
Cognitive improvement effects of PF-04957325, a phosphodiesterase-8 inhibitor, in mouse models of Alzheimer's disease via modulating neuroinflammation.Int J Neuropsychopharmacol. 2025 May 9;28(5):pyaf028. doi: 10.1093/ijnp/pyaf028. Int J Neuropsychopharmacol. 2025. PMID: 40312965 Free PMC article.
-
Infections and immunity: associations with obesity and related metabolic disorders.J Pathol Transl Med. 2023 Jan;57(1):28-42. doi: 10.4132/jptm.2022.11.14. Epub 2023 Jan 15. J Pathol Transl Med. 2023. PMID: 36647284 Free PMC article. Review.
-
Effects of Prion Protein on Aβ42 and Pyroglutamate-Modified AβpΕ3-42 Oligomerization and Toxicity.Mol Neurobiol. 2019 Mar;56(3):1957-1971. doi: 10.1007/s12035-018-1202-x. Epub 2018 Jul 6. Mol Neurobiol. 2019. PMID: 29981054
-
Manipulating the Prion Protein Gene Sequence and Expression Levels with CRISPR/Cas9.PLoS One. 2016 Apr 29;11(4):e0154604. doi: 10.1371/journal.pone.0154604. eCollection 2016. PLoS One. 2016. PMID: 27128441 Free PMC article.
-
Molecular Mechanisms Associated with Neurodegeneration of Neurotropic Viral Infection.Mol Neurobiol. 2024 May;61(5):2881-2903. doi: 10.1007/s12035-023-03761-6. Epub 2023 Nov 9. Mol Neurobiol. 2024. PMID: 37946006 Free PMC article. Review.
References
-
- Querfurth HW, LaFerla FM. Alzheimer's disease. N Engl J Med. 2010; 362:329-344 - PubMed
-
- Berchtold NC, Cotman CW. Evolution in the conceptualization of dementia and Alzheimer's disease: Greco-Roman period to the 1960s. Neurobiol Aging. 1998; 19:173-189 - PubMed
-
- Maurer K, Volk S, Gerbaldo H. Auguste D and Alzheimer's disease. Lancet. 1997; 349:1546-1549 - PubMed
-
- Alzheimer A. About a peculiar disease of the cerebral cortex. Centralblatt fur Nervenheilkunde Psychiatrie. 1907; 30:177-179 (in German)
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous