

cAMP controls the restoration of endothelial barrier function after thrombin-induced hyperpermeability via Rac1 activation
- PMID: 25344477
- PMCID: PMC4254100
- DOI: 10.14814/phy2.12175
cAMP controls the restoration of endothelial barrier function after thrombin-induced hyperpermeability via Rac1 activation
Abstract
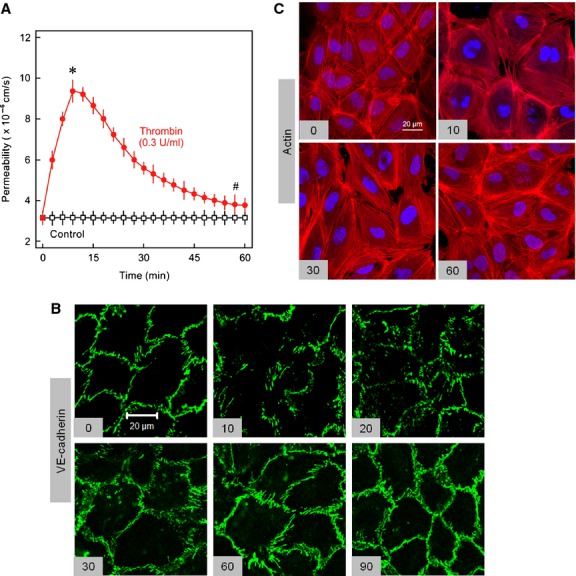
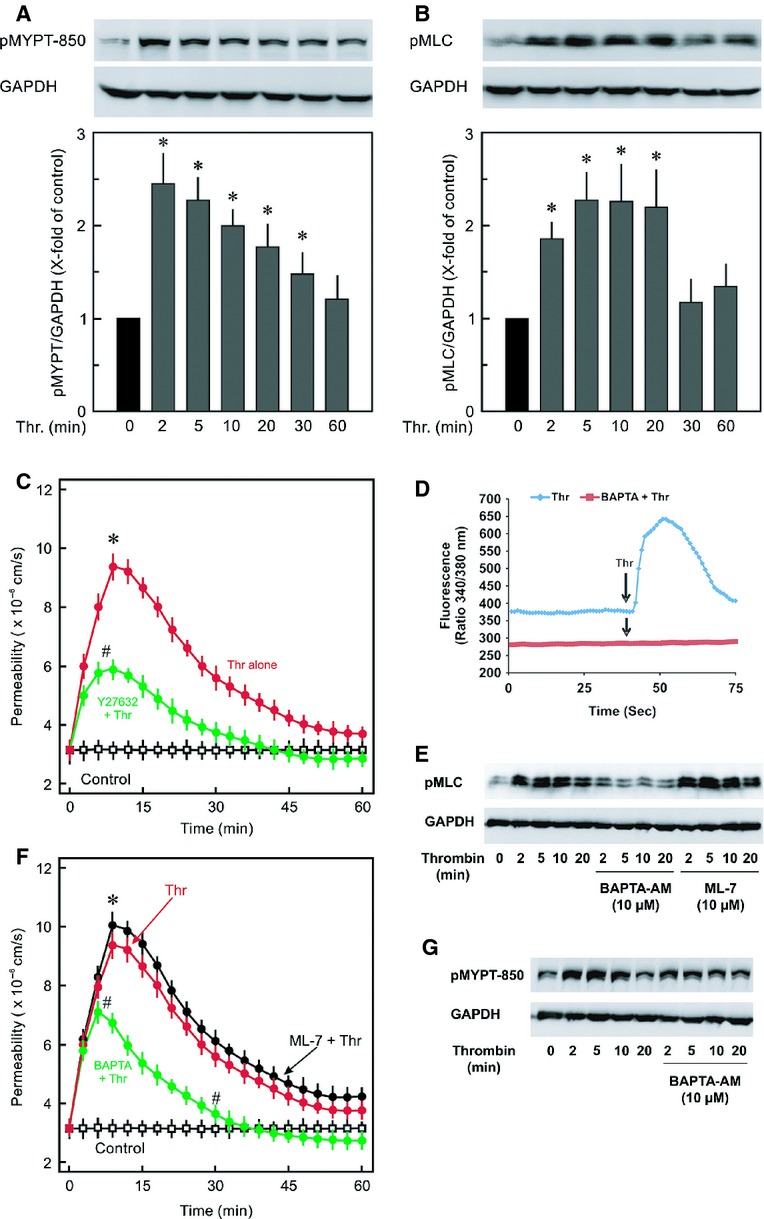
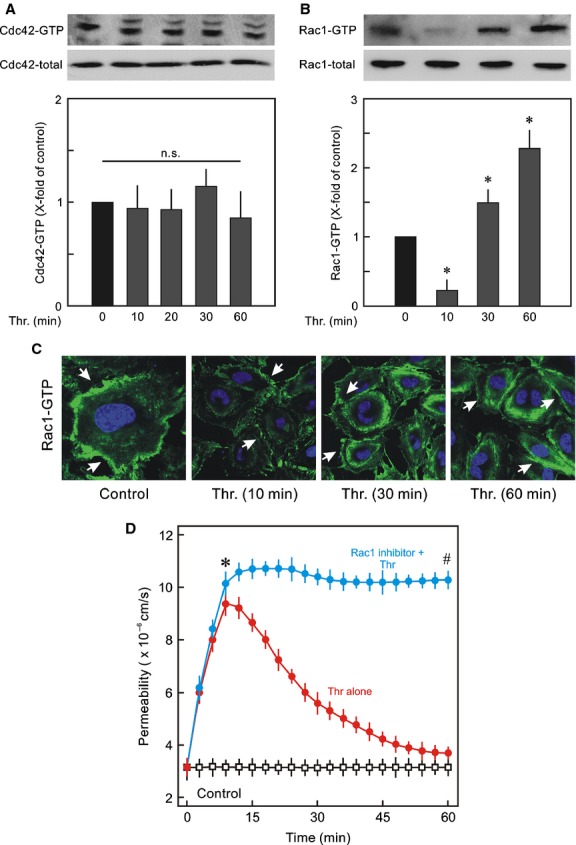
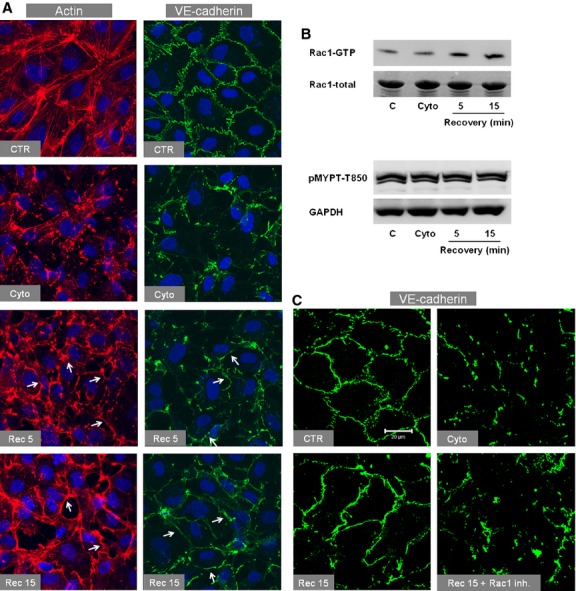
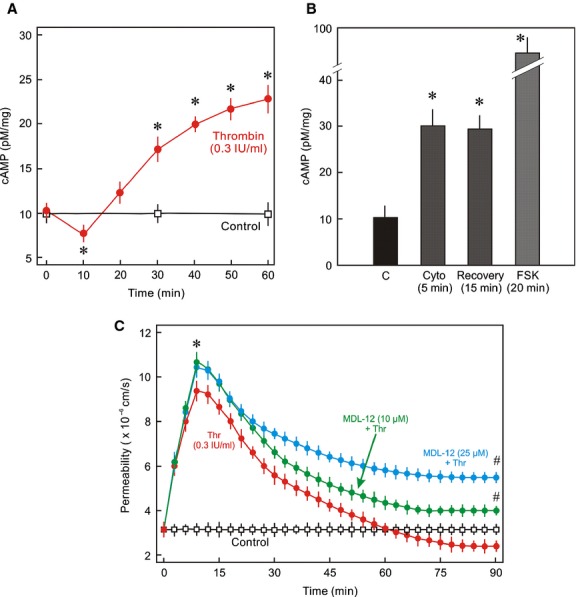
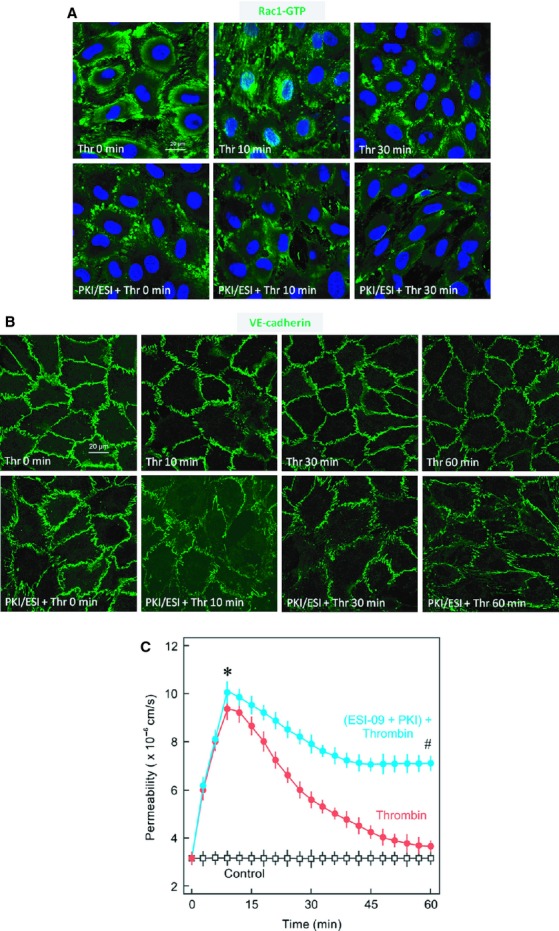
Inflammatory mediators like thrombin disrupt endothelial adherens junctions (AJs) and barrier integrity leading to oedema formation followed by resealing of AJs and a slow recovery of the barrier function. The molecular mechanisms of this process have not yet been fully delineated. The aim of the present study was to analyse the molecular mechanism of endothelial barrier recovery and thrombin was used as model inflammatory mediator. Thrombin caused a strong increase in endothelial permeability within 10 min accompanied by loss of Rac1 but not cdc42 activity, drop in cellular cAMP contents, and a strong activation of the endothelial contractile machinery mainly via RhoA/Rock signalling. Activation of RhoA/Rock signalling precedes and is dependent upon a rise in the cytosolic Ca(2+) concentration. Inhibition of cytosolic Ca(2+) rise but not MLCK or Rock enhances the recovery of endothelial barrier function. The cellular cAMP contents increased gradually during the barrier recovery phase (30-60 min after thrombin challenge) accompanied by an increase in Rac1 activity. Inhibition of Rac1 activity using a specific pharmacological inhibitor (NSC23766) abrogated the endothelial barrier recovery process, suggesting a Rac1-dependent phenomenon. Likewise, inhibition of either adenylyl cyclase or the cAMP-effectors PKA and Epac (with PKI and ESI-09, respectively) caused an abrogation of Rac1 activation, resealing of endothelial AJs and recovery of endothelial barrier function. The data demonstrate that endothelial barrier recovery after thrombin challenge is regulated by Rac1 GTPase activation. This Rac1 activation is due to increased levels of cellular cAMP and activation of downstream signalling during the barrier recovery phase.
Keywords: Adenylyl cyclase; Rac1; RhoA; adherens junctions; calcium.
© 2014 The Authors. Physiological Reports published by Wiley Periodicals, Inc. on behalf of the American Physiological Society and The Physiological Society.
Figures
References
-
- Adamson R. H., Zeng M., Adamson G. N., Lenz J. F., Curry F. E. 2003. PAF‐ and bradykinin‐induced hyperpermeability of rat venules is independent of actin‐myosin contraction. Am. J. Physiol. Heart Circ. Physiol.; 285:H406-H417. - PubMed
-
- Aslam M., Härtel F. V., Arshad M., Gündüz D., Abdallah Y., Sauer H. 2010. cAMP/PKA antagonizes thrombin‐induced inactivation of endothelial myosin light chain phosphatase: Role of CPI‐17. Cardiovasc. Res.; 87:375-384. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
