

Small RNAs from plants, bacteria and fungi within the order Hypocreales are ubiquitous in human plasma
- PMID: 25344700
- PMCID: PMC4230795
- DOI: 10.1186/1471-2164-15-933
Small RNAs from plants, bacteria and fungi within the order Hypocreales are ubiquitous in human plasma
Abstract
Background: The human microbiome plays a significant role in maintaining normal physiology. Changes in its composition have been associated with bowel disease, metabolic disorders and atherosclerosis. Sequences of microbial origin have been observed within small RNA sequencing data obtained from blood samples. The aim of this study was to characterise the microbiome from which these sequences are derived.
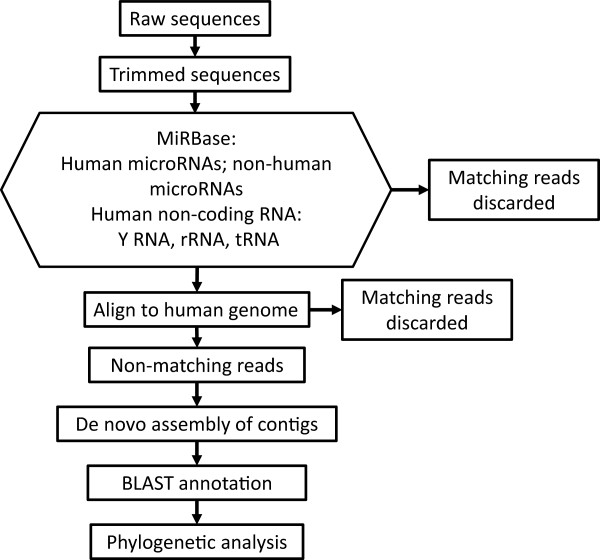
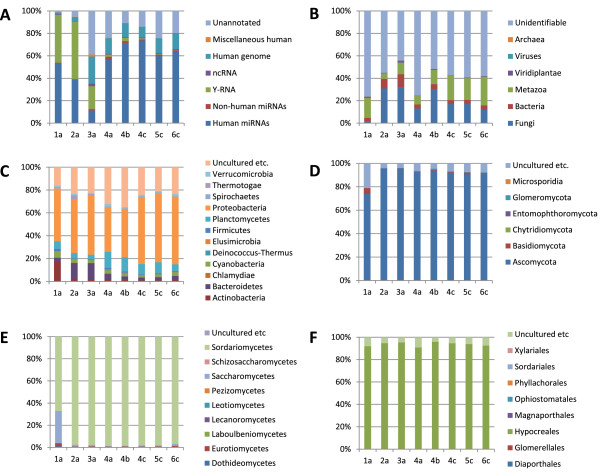
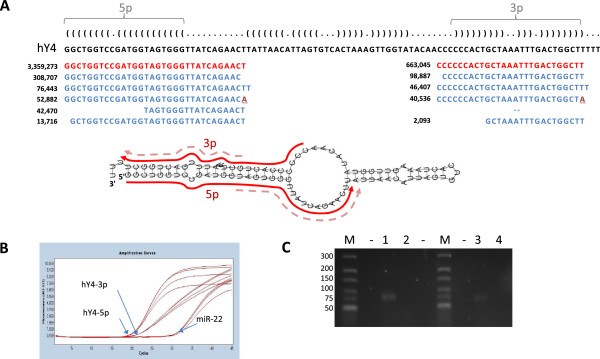
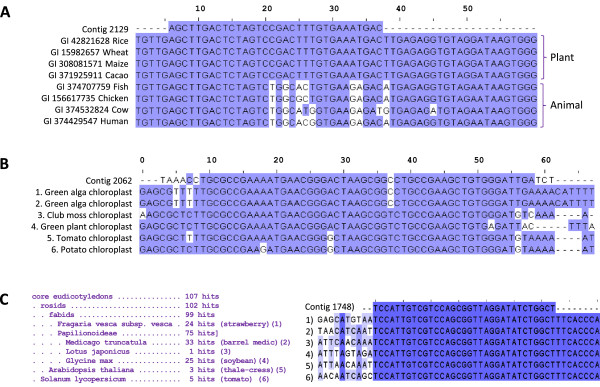
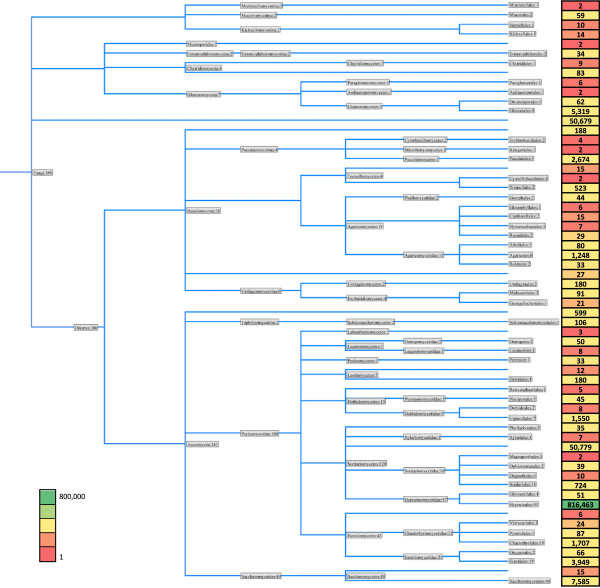
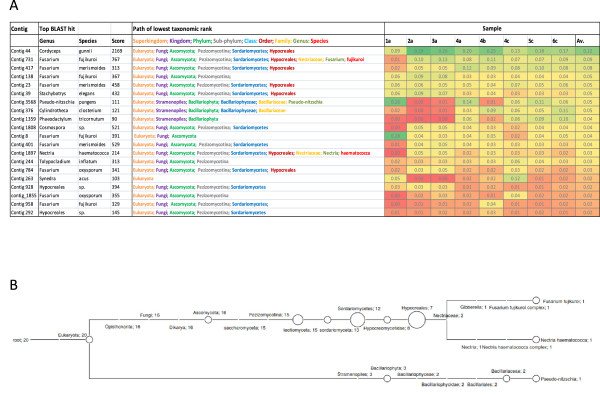
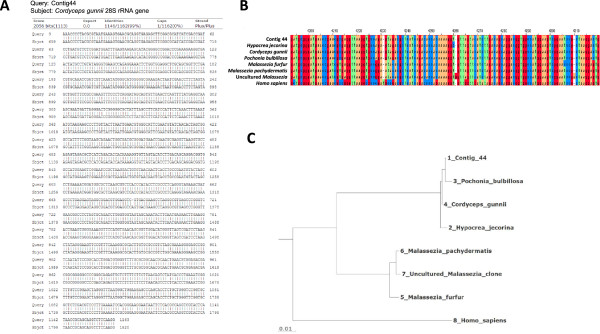
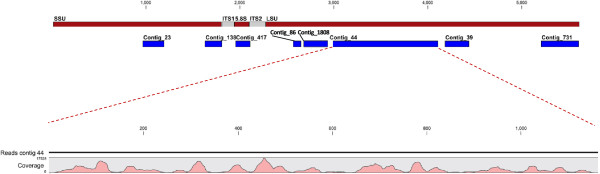
Results: Abundant non-human small RNA sequences were identified in plasma and plasma exosomal samples. Assembly of these short sequences into longer contigs was the pivotal novel step in ascertaining their origin by BLAST searches. Most reads mapped to rRNA sequences. The taxonomic profiles of the microbes detected were very consistent between individuals but distinct from microbiomes reported at other sites. The majority of bacterial reads were from the phylum Proteobacteria, whilst for 5 of 6 individuals over 90% of the more abundant fungal reads were from the phylum Ascomycota; of these over 90% were from the order Hypocreales. Many contigs were from plants, presumably of dietary origin. In addition, extremely abundant small RNAs derived from human Y RNAs were detected.
Conclusions: A characteristic profile of a subset of the human microbiome can be obtained by sequencing small RNAs present in the blood. The source and functions of these molecules remain to be determined, but the specific profiles are likely to reflect health status. The potential to provide biomarkers of diet and for the diagnosis and prognosis of human disease is immense.
Figures
References
-
- Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, Mende DR, Li J, Xu J, Li S, Li D, Cao J, Wang B, Liang H, Zheng H, Xie Y, Tap J, Lepage P, Bertalan M, Batto JM, Hansen T, Le Paslier D, Linneberg A, Nielsen HB, Pelletier E, Renault P, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464(7285):59–65. - PMC - PubMed
-
- Human Microbiome Project (HMP)http://commonfund.nih.gov/Hmp/
-
- Ott SJ, Kuhbacher T, Musfeldt M, Rosenstiel P, Hellmig S, Rehman A, Drews O, Weichert W, Timmis KN, Schreiber S. Fungi and inflammatory bowel diseases: Alterations of composition and diversity. Scand J Gastroenterol. 2008;43(7):831–841. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
