

Genotype-by-sequencing of the plant-pathogenic fungi Pyrenophora teres and Sphaerulina musiva utilizing Ion Torrent sequence technology
- PMID: 25346350
- PMCID: PMC6638358
- DOI: 10.1111/mpp.12214
Genotype-by-sequencing of the plant-pathogenic fungi Pyrenophora teres and Sphaerulina musiva utilizing Ion Torrent sequence technology
Abstract
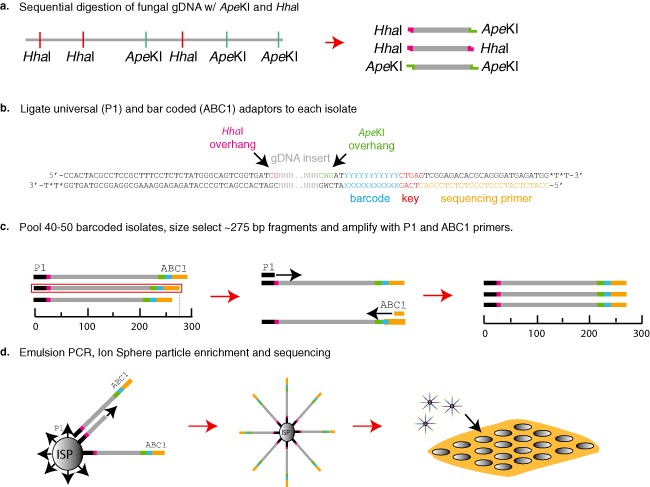
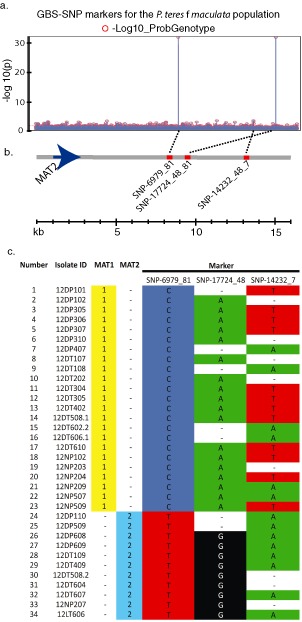
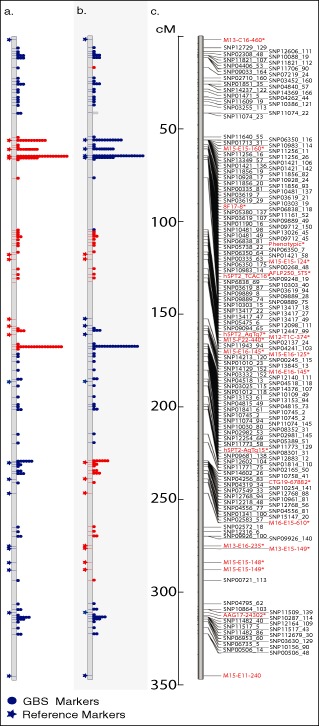
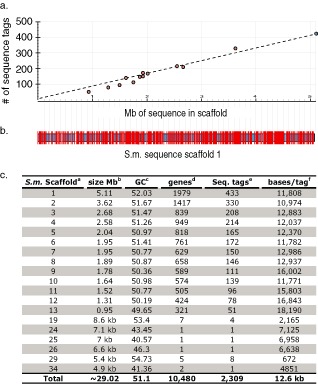
Genetic and genomics tools to characterize host-pathogen interactions are disproportionately directed to the host because of the focus on resistance. However, understanding the genetics of pathogen virulence is equally important and has been limited by the high cost of de novo genotyping of species with limited marker data. Non-resource-prohibitive methods that overcome the limitation of genotyping are now available through genotype-by-sequencing (GBS). The use of a two-enzyme restriction-associated DNA (RAD)-GBS method adapted for Ion Torrent sequencing technology provided robust and reproducible high-density genotyping of several fungal species. A total of 5783 and 2373 unique loci, 'sequence tags', containing 16,441 and 9992 single nucleotide polymorphisms (SNPs) were identified and characterized from natural populations of Pyrenophora teres f. maculata and Sphaerulina musiva, respectively. The data generated from the P. teres f. maculata natural population were used in association mapping analysis to map the mating-type gene to high resolution. To further validate the methodology, a biparental population of P. teres f. teres, previously used to develop a genetic map utilizing simple sequence repeat (SSR) and amplified fragment length polymorphism (AFLP) markers, was re-analysed using the SNP markers generated from this protocol. A robust genetic map containing 1393 SNPs on 997 sequence tags spread across 15 linkage groups with anchored reference markers was generated from the P. teres f. teres biparental population. The robust high-density markers generated using this protocol will allow positional cloning in biparental fungal populations, association mapping of natural fungal populations and population genetics studies.
Keywords: Pyrenophora teres; Sphaerulina musiva; fungi; genotype-by-sequencing.
© 2014 BSPP AND JOHN WILEY & SONS LTD.
Figures

References
-
- Antequera, F. , Tamame, M. , Villanueva, J.R. and Santos, T. (1984) DNA methylation in the fungi. J. Biol. Chem. 259, 8033–8036. - PubMed
-
- Arumuganathan, K. and Earle, E. (1991) Nuclear DNA content of some important plant species. Plant Mol. Biol. Rep. 9, 208–218.
-
- Binz, T. , D'Mello, N. and Horgen, P.A. (1998) A comparison of DNA methylation levels in selected isolates of higher fungi. Mycologia, 90, 785–790.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
