

A single-gene cause in 29.5% of cases of steroid-resistant nephrotic syndrome
- PMID: 25349199
- PMCID: PMC4446877
- DOI: 10.1681/ASN.2014050489
A single-gene cause in 29.5% of cases of steroid-resistant nephrotic syndrome
Abstract
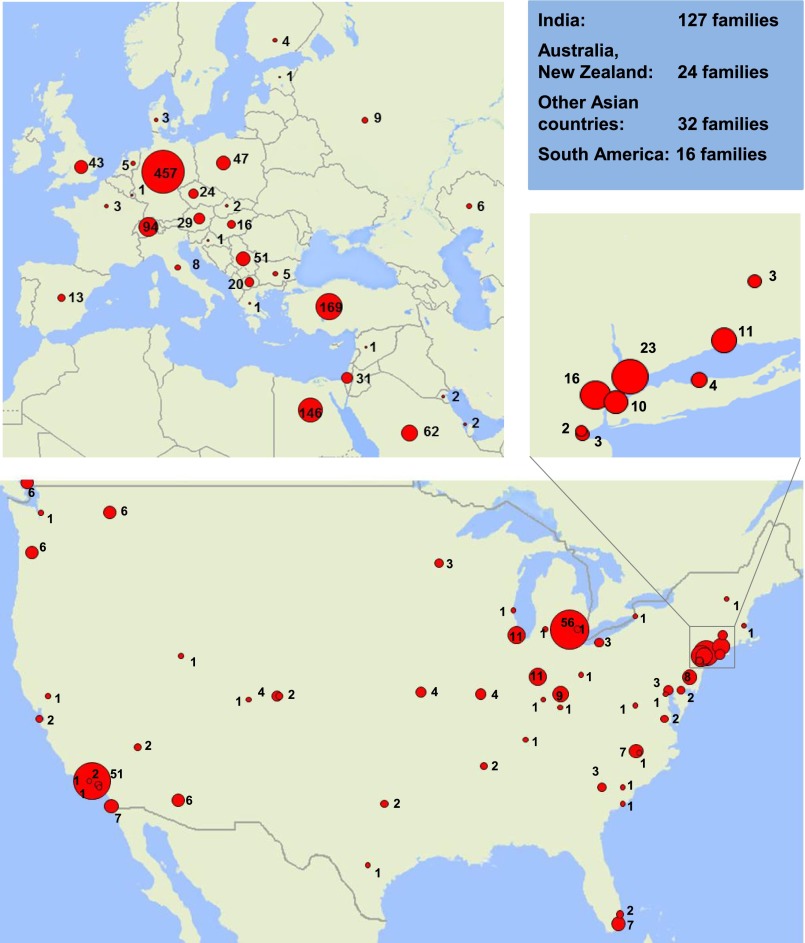
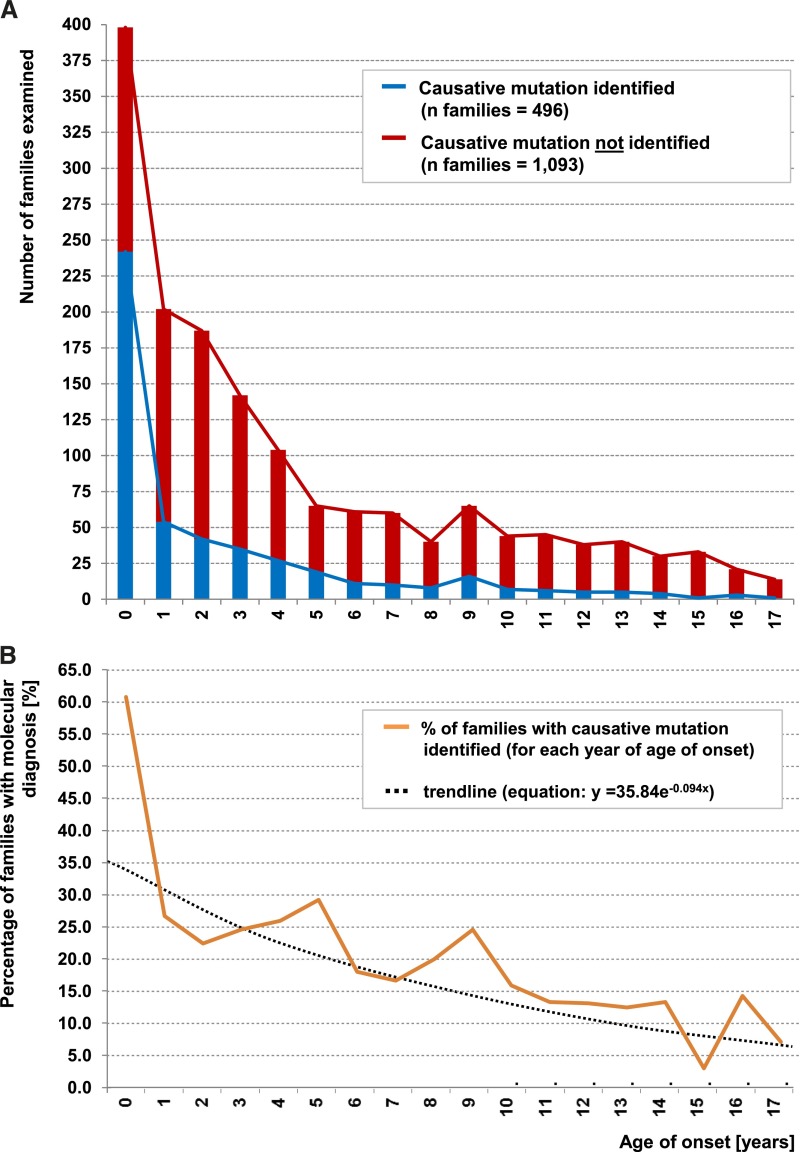
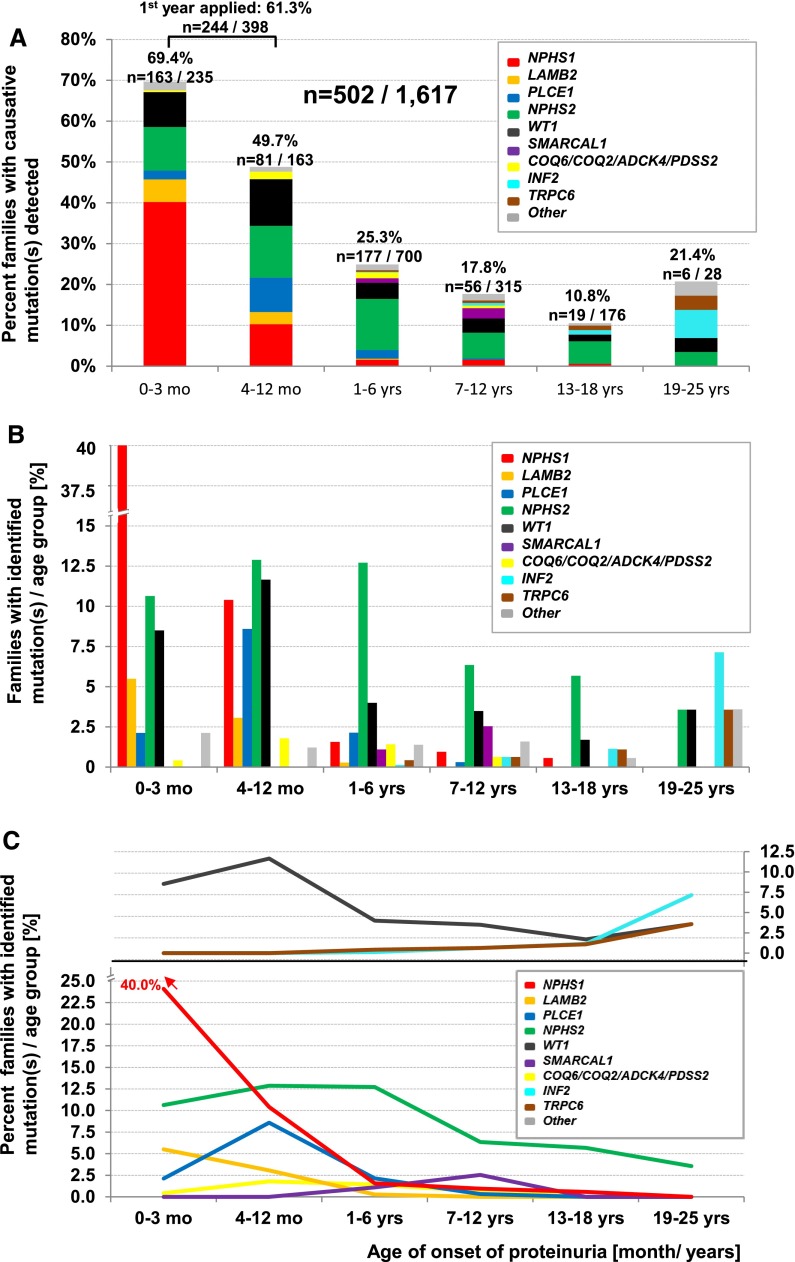
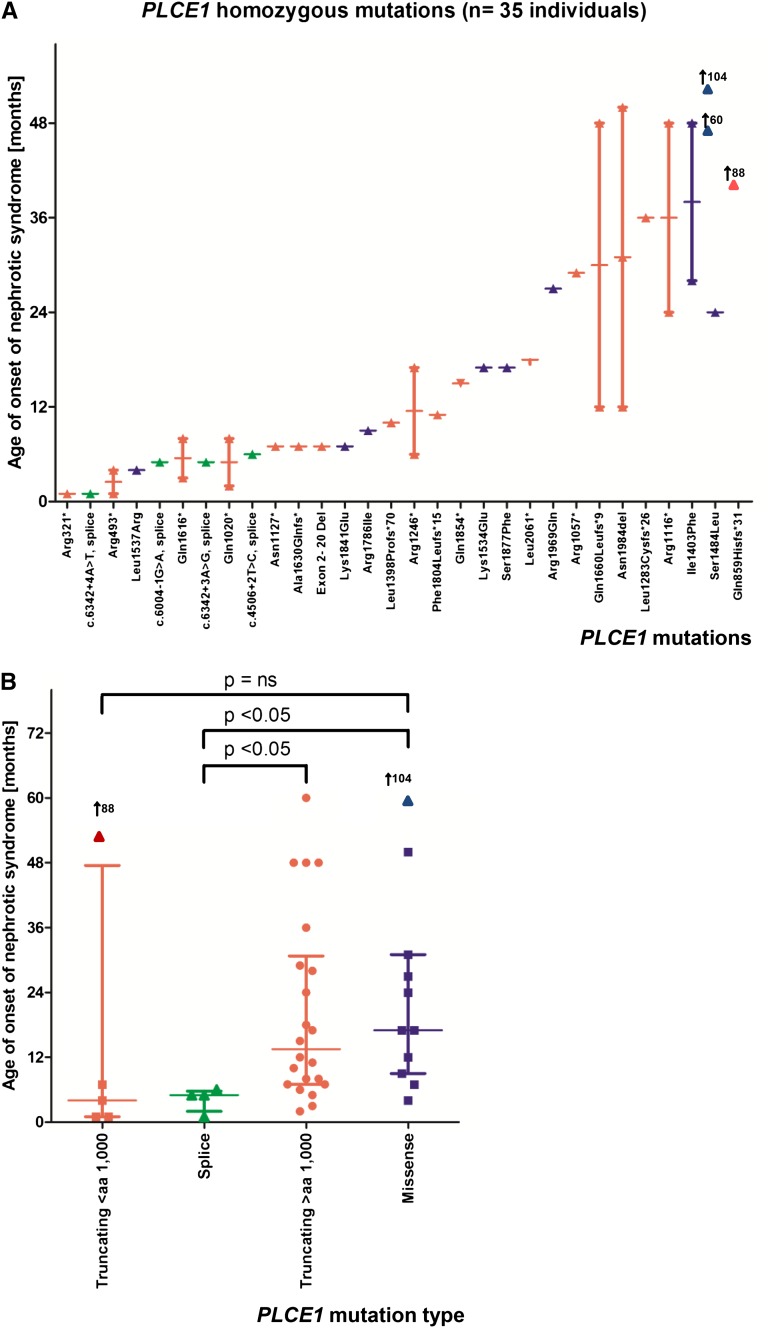
Steroid-resistant nephrotic syndrome (SRNS) is the second most frequent cause of ESRD in the first two decades of life. Effective treatment is lacking. First insights into disease mechanisms came from identification of single-gene causes of SRNS. However, the frequency of single-gene causation and its age distribution in large cohorts are unknown. We performed exon sequencing of NPHS2 and WT1 for 1783 unrelated, international families with SRNS. We then examined all patients by microfluidic multiplex PCR and next-generation sequencing for all 27 genes known to cause SRNS if mutated. We detected a single-gene cause in 29.5% (526 of 1783) of families with SRNS that manifested before 25 years of age. The fraction of families in whom a single-gene cause was identified inversely correlated with age of onset. Within clinically relevant age groups, the fraction of families with detection of the single-gene cause was as follows: onset in the first 3 months of life (69.4%), between 4 and 12 months old (49.7%), between 1 and 6 years old (25.3%), between 7 and 12 years old (17.8%), and between 13 and 18 years old (10.8%). For PLCE1, specific mutations correlated with age of onset. Notably, 1% of individuals carried mutations in genes that function within the coenzyme Q10 biosynthesis pathway, suggesting that SRNS may be treatable in these individuals. Our study results should facilitate molecular genetic diagnostics of SRNS, etiologic classification for therapeutic studies, generation of genotype-phenotype correlations, and the identification of individuals in whom a targeted treatment for SRNS may be available.
Keywords: FSGS; SRNS; genetic disease; kidney failure; nephrosis; steroid-resistant nephrotic syndrome.
Copyright © 2015 by the American Society of Nephrology.
Figures
References
-
- Smith JM, Stablein DM, Munoz R, Hebert D, McDonald RA: Contributions of the Transplant Registry: The 2006 Annual Report of the North American Pediatric Renal Trials and Collaborative Studies (NAPRTCS). Pediatr Transplant 11: 366–373, 2007 - PubMed
-
- Cheong HI, Han HW, Park HW, Ha IS, Han KS, Lee HS, Kim SJ, Choi Y. Early recurrent nephrotic syndrome after renal transplantation in children with focal segmental glomerulosclerosis. Nephrol Dial Transplant 15: 78-81, 2000 - PubMed
-
- Senggutuvan P, Cameron JS, Hartley RB, Rigden S, Chantler C, Haycock G, Williams DG, Ogg C, Koffman G: Recurrence of focal segmental glomerulosclerosis in transplanted kidneys: Analysis of incidence and risk factors in 59 allografts. Pediatr Nephrol 4: 21–28, 1990 - PubMed
-
- Ruf RG, Lichtenberger A, Karle SM, Haas JP, Anacleto FE, Schultheiss M, Zalewski I, Imm A, Ruf EM, Mucha B, Bagga A, Neuhaus T, Fuchshuber A, Bakkaloglu A, Hildebrandt F, Arbeitsgemeinschaft Für Pädiatrische Nephrologie Study Group : Patients with mutations in NPHS2 (podocin) do not respond to standard steroid treatment of nephrotic syndrome. J Am Soc Nephrol 15: 722–732, 2004 - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
