

Incomplete penetrance and phenotypic variability of 6q16 deletions including SIM1
- PMID: 25351778
- PMCID: PMC4795105
- DOI: 10.1038/ejhg.2014.230
Incomplete penetrance and phenotypic variability of 6q16 deletions including SIM1
Abstract
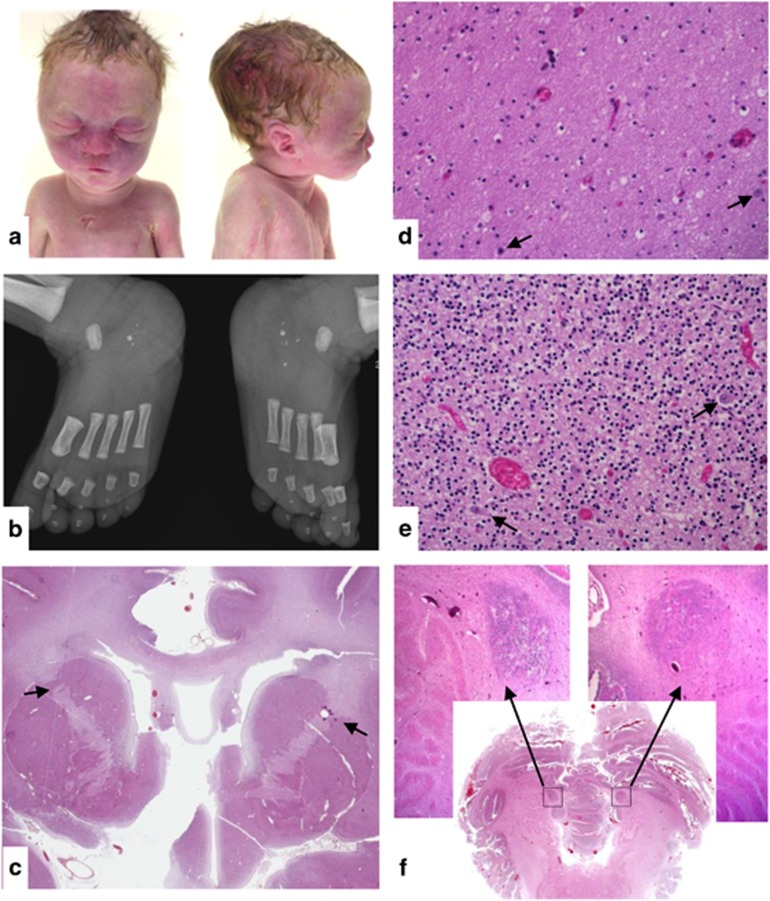
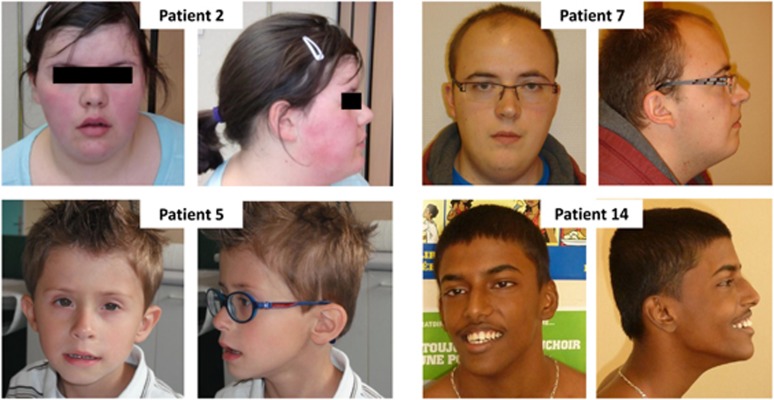
6q16 deletions have been described in patients with a Prader-Willi-like (PWS-like) phenotype. Recent studies have shown that certain rare single-minded 1 (SIM1) loss-of-function variants were associated with a high intra-familial risk for obesity with or without features of PWS-like syndrome. Although SIM1 seems to have a key role in the phenotype of patients carrying 6q16 deletions, some data support a contribution of other genes, such as GRIK2, to explain associated behavioural problems. We describe 15 new patients in whom de novo 6q16 deletions were characterised by comparative genomic hybridisation or single-nucleotide polymorphism (SNP) array analysis, including the first patient with fetopathological data. This fetus showed dysmorphic facial features, cerebellar and cerebral migration defects with neuronal heterotopias, and fusion of brain nuclei. The size of the deletion in the 14 living patients ranged from 1.73 to 7.84 Mb, and the fetus had the largest deletion (14 Mb). Genotype-phenotype correlations confirmed the major role for SIM1 haploinsufficiency in obesity and the PWS-like phenotype. Nevertheless, only 8 of 13 patients with SIM1 deletion exhibited obesity, in agreement with incomplete penetrance of SIM1 haploinsufficiency. This study in the largest series reported to date confirms that the PWS-like phenotype is strongly linked to 6q16.2q16.3 deletions and varies considerably in its clinical expression. The possible involvement of other genes in the 6q16.2q16.3-deletion phenotype is discussed.
Figures

References
-
- Cassidy SB, Schwartz S, Miller JL, Driscoll DJ: Prader-Willi syndrome. GenetMed 2012; 14: 10–26. - PubMed
-
- D'Angelo CS, Kohl I, Varela MC et al: Obesity with associated developmental delay and/or learning disability in patients exhibiting additional features: report of novel pathogenic copy number variants. Am J Med Genet A 2013; 161A: 479–486. - PubMed
-
- Cox H, Bullman H, Temple IK: Maternal UPD(14) in the patient with a normal karyotype: clinical report and a systematic search for cases in samples sent for testing for Prader-Willi syndrome. Am J Med Genet A 2004; 127A: 21–25. - PubMed
-
- Hosoki K, Kagami M, Tanaka T et al: Maternal uniparental disomy 14 syndrome demonstrates prader-willi syndrome-like phenotype. J Pediatr 2009; 155: 900–903, e901. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
