

Severe retinal degeneration in women with a c.2543del mutation in ORF15 of the RPGR gene
- PMID: 25352739
- PMCID: PMC4169777
Severe retinal degeneration in women with a c.2543del mutation in ORF15 of the RPGR gene
Abstract
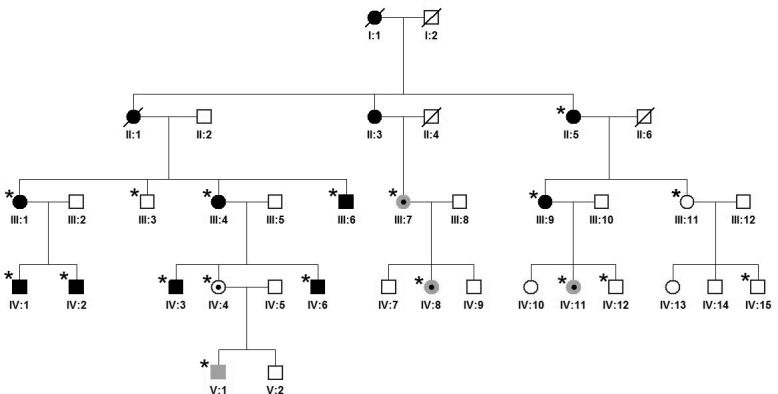
Purpose: To describe the genotype-phenotype correlation and serial observations in a five-generation Czech family with X-linked retinitis pigmentosa (XLRP) associated with severe visual impairment in women.
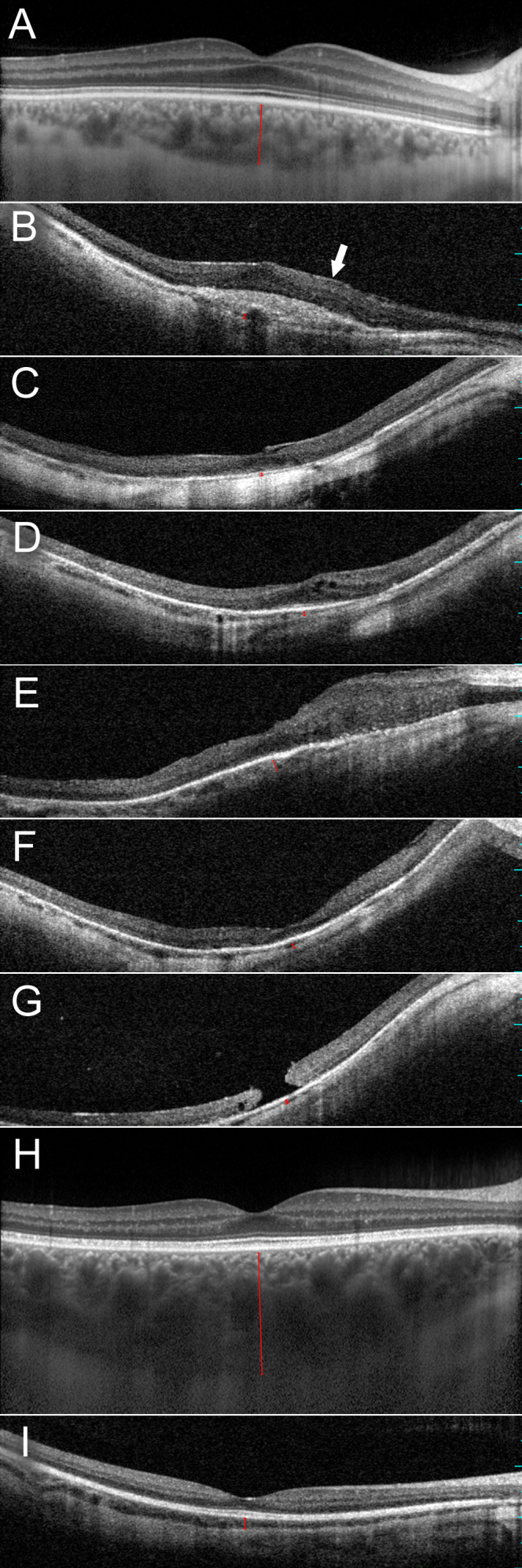
Methods: Comprehensive ophthalmological examination including spectral domain optical coherence tomography (SD-OCT) was performed. Based on the pedigree structure and women being severely affected, autosomal dominant inheritance was suspected, and screening for known mutations by genotyping microarray was performed. Subsequently, direct sequencing of ORF15 RPGR was undertaken.
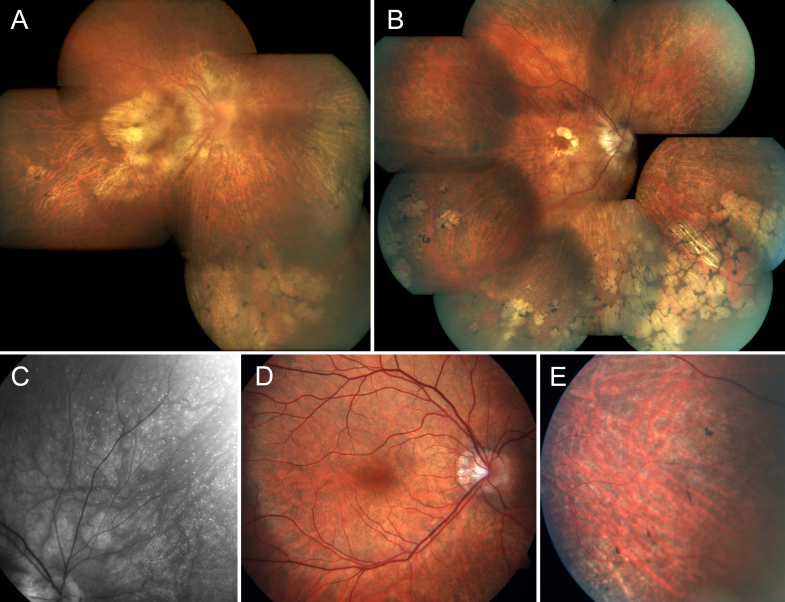
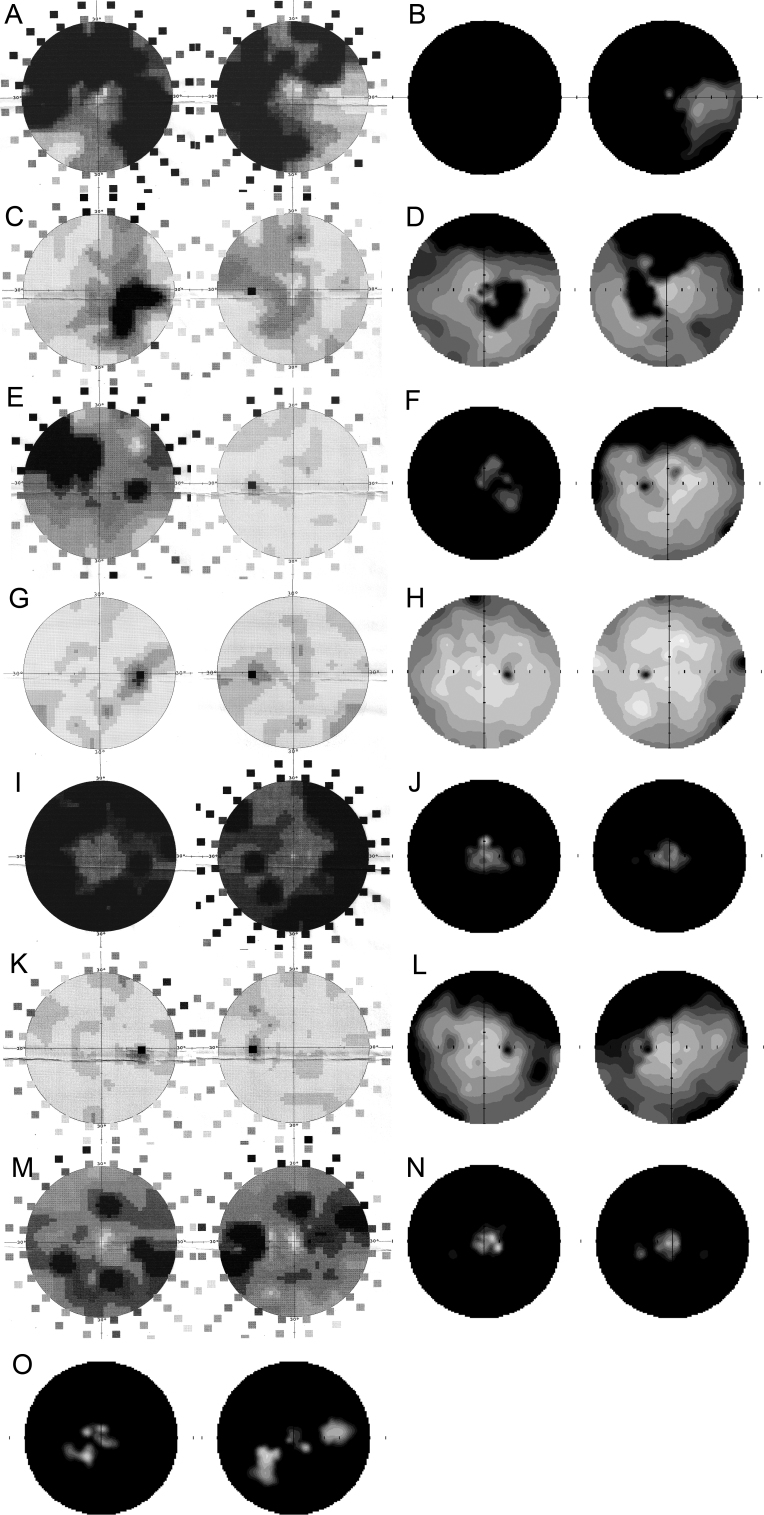
Results: Eighteen family members (nine women and nine men) were examined. A pathogenic variant, c.2543del in ORF15 of RPGR, was found to segregate with disease. The oldest woman and her two sisters had no perception of light in their sixth decade. Four women and five men had signs and symptoms of typical XLRP, including moderate to high myopia. Three other women also had moderate to high myopia and myopic astigmatism but without the presence of bone spicule-like formation. Severe disruption of macular architecture on SD-OCT was equally common in both sexes. Only one 32-year-old female carrier had clinically normal findings. Subfoveal choroidal thickness was decreased in all affected men and in all female carriers, except the only carrier with a normal fundus examination.
Conclusions: The c.2543del mutation in ORF15 of RPGR is associated with a severe phenotype in the women in this family. The presence of a significant myopic refractive error, in the absence of male-to-male transmission, may be indicative of X-linked inheritance. Measurements of choroidal thickness may help in clinically identifying carrier status.
Figures
References
-
- Berger W, Kloeckener-Gruissem B, Neidhardt J. The molecular basis of human retinal and vitreoretinal diseases. Prog Retin Eye Res. 2010;29:335–75. - PubMed
-
- Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368:1795–809. - PubMed
-
- Koenekoop RK, Loyer M, Hand CK, Al Mahdi H, Dembinska O, Beneish R, Racine J, Rouleau GA. Novel RPGR mutations with distinct retinitis pigmentosa phenotypes in French-Canadian families. Am J Ophthalmol. 2003;136:678–87. - PubMed
-
- Banin E, Mizrahi-Meissonnier L, Neis R, Silverstein S, Magyar I, Abeliovich D, Roepman R, Berger W, Rosenberg T, Sharon D. A non-ancestral RPGR missense mutation in families with either recessive or semi-dominant X-linked retinitis pigmentosa. Am J Med Genet A. 2007;143A:1150–8. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
