

Diverse and widespread contamination evident in the unmapped depths of high throughput sequencing data
- PMID: 25354084
- PMCID: PMC4213012
- DOI: 10.1371/journal.pone.0110808
Diverse and widespread contamination evident in the unmapped depths of high throughput sequencing data
Abstract
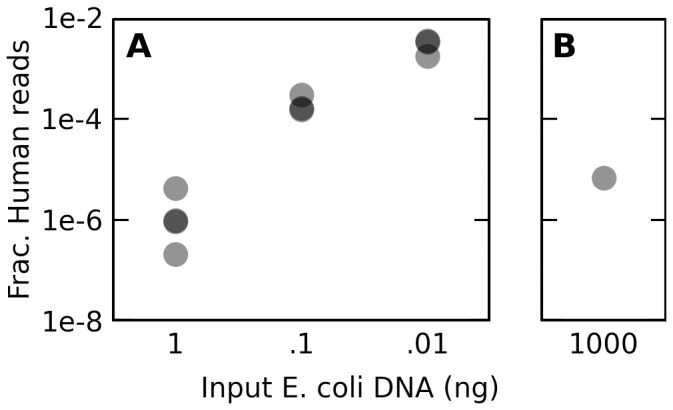
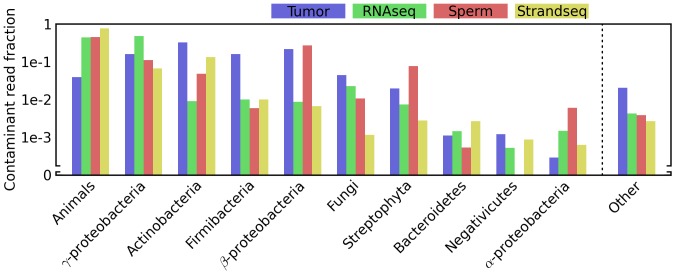
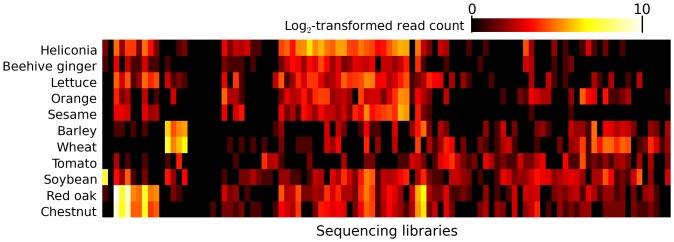
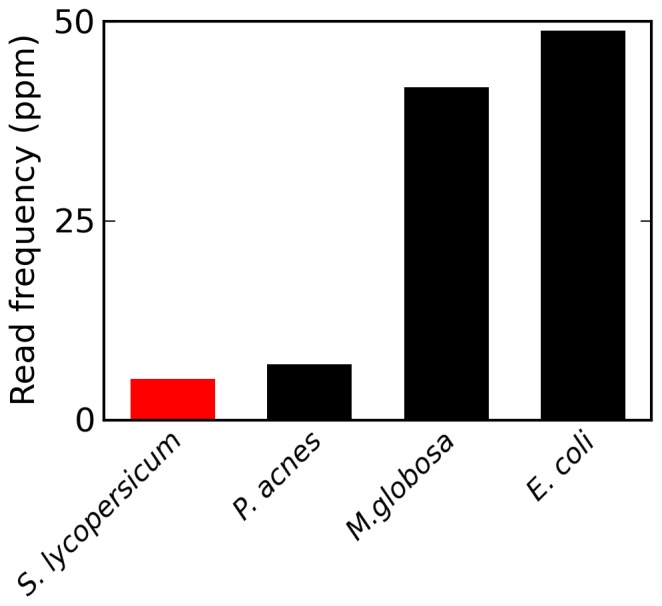
Trace quantities of contaminating DNA are widespread in the laboratory environment, but their presence has received little attention in the context of high throughput sequencing. This issue is highlighted by recent works that have rested controversial claims upon sequencing data that appear to support the presence of unexpected exogenous species. I used reads that preferentially aligned to alternate genomes to infer the distribution of potential contaminant species in a set of independent sequencing experiments. I confirmed that dilute samples are more exposed to contaminating DNA, and, focusing on four single-cell sequencing experiments, found that these contaminants appear to originate from a wide diversity of clades. Although negative control libraries prepared from 'blank' samples recovered the highest-frequency contaminants, low-frequency contaminants, which appeared to make heterogeneous contributions to samples prepared in parallel within a single experiment, were not well controlled for. I used these results to show that, despite heavy replication and plausible controls, contamination can explain all of the observations used to support a recent claim that complete genes pass from food to human blood. Contamination must be considered a potential source of signals of exogenous species in sequencing data, even if these signals are replicated in independent experiments, vary across conditions, or indicate a species which seems a priori unlikely to contaminate. Negative control libraries processed in parallel are essential to control for contaminant DNAs, but their limited ability to recover low-frequency contaminants must be recognized.
Conflict of interest statement
Figures

References
-
- Schmidt T, Hummel S, Herrmann B (1995) Evidence of contamination in PCR laboratory disposables. Naturwissenschaften 82(9): 423–31. - PubMed
-
- Leonard JA, Shanks O, Hofreiter M, Kreuz E, Hodges L, et al. (2007) Animal DNA in PCR reagents plagues ancient DNA research. Journal of Archaeological Science 34(9): 1361–6.
-
- Peters RP, Mohammadi T, Vandenbroucke Grauls CM, Danner SA, van Agtmael MA, et al. (2004) Detection of bacterial DNA in blood samples from febrile patients: underestimated infection or emerging contamination? FEMS Immunol Med Microbiol 42(2): 249–53. - PubMed
-
- Ehricht R, Hotzel H, Sachse K, Slickers P (2007) Residual DNA in thermostable DNA polymerases - a cause of irritation in diagnostic PCR and microarray assays. Biologicals 35(2): 145–7. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
