

Computational modeling of membrane proteins
- PMID: 25355688
- PMCID: PMC4270820
- DOI: 10.1002/prot.24703
Computational modeling of membrane proteins
Abstract
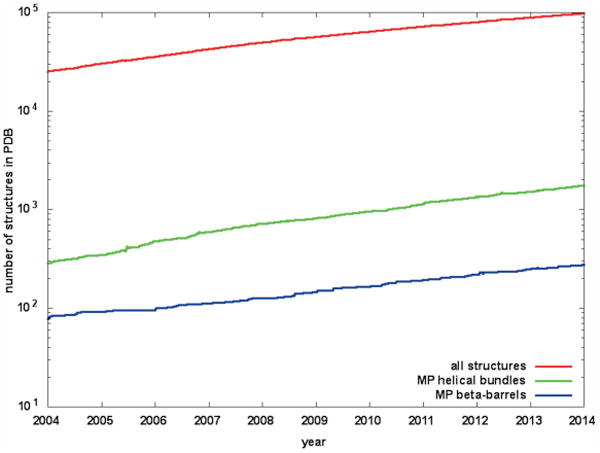
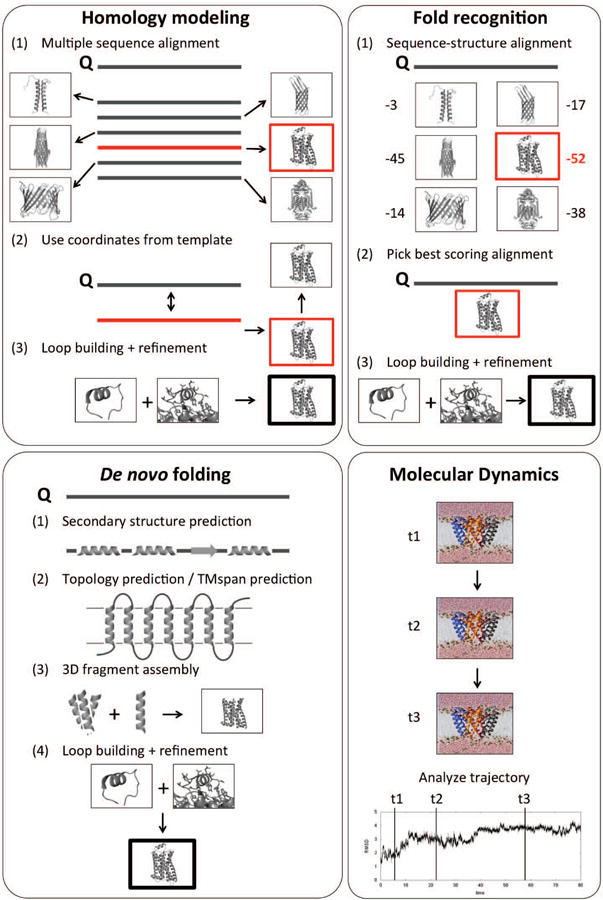
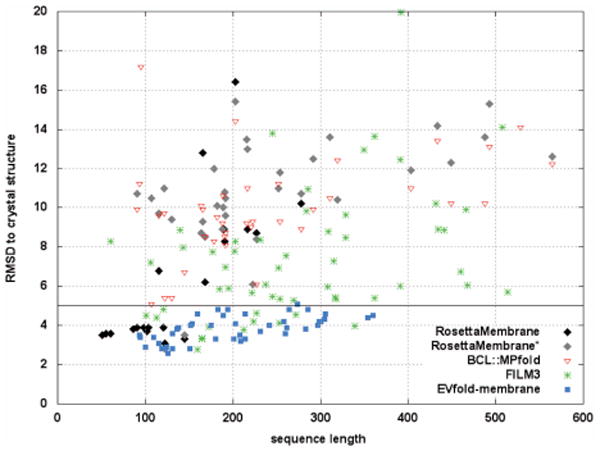
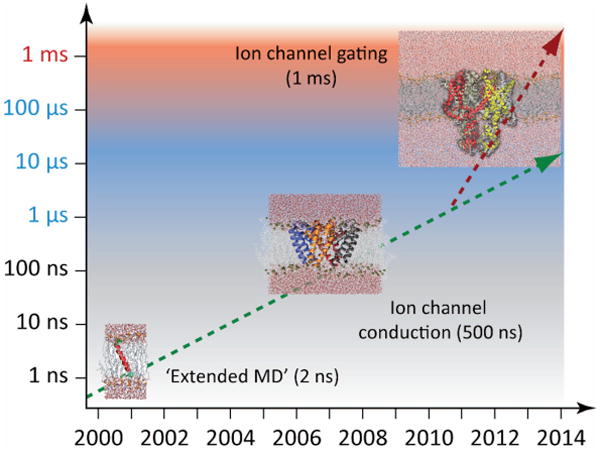
The determination of membrane protein (MP) structures has always trailed that of soluble proteins due to difficulties in their overexpression, reconstitution into membrane mimetics, and subsequent structure determination. The percentage of MP structures in the protein databank (PDB) has been at a constant 1-2% for the last decade. In contrast, over half of all drugs target MPs, only highlighting how little we understand about drug-specific effects in the human body. To reduce this gap, researchers have attempted to predict structural features of MPs even before the first structure was experimentally elucidated. In this review, we present current computational methods to predict MP structure, starting with secondary structure prediction, prediction of trans-membrane spans, and topology. Even though these methods generate reliable predictions, challenges such as predicting kinks or precise beginnings and ends of secondary structure elements are still waiting to be addressed. We describe recent developments in the prediction of 3D structures of both α-helical MPs as well as β-barrels using comparative modeling techniques, de novo methods, and molecular dynamics (MD) simulations. The increase of MP structures has (1) facilitated comparative modeling due to availability of more and better templates, and (2) improved the statistics for knowledge-based scoring functions. Moreover, de novo methods have benefited from the use of correlated mutations as restraints. Finally, we outline current advances that will likely shape the field in the forthcoming decade.
Keywords: alpha-helical membrane proteins; beta-barrel membrane proteins; de novo folding; homology modeling; membrane proteins; molecular dynamics simulations; protein modeling; protein structure; sequence-based methods; structure prediction.
© 2014 Wiley Periodicals, Inc.
Conflict of interest statement
Figures
References
-
- Jones DT. Do transmembrane protein superfolds exist? FEBS Lett. 1998;423:281–285. - PubMed
-
- Yildirim MA, Goh KI, Cusick ME, Barabási AL, Vidal M. Drug-target network. Nat Biotechnol. 2007;25:1119–1126. - PubMed
-
- Grisshammer R, Tate CG. Overexpression of integral membrane proteins for structural studies. Q Rev Biophys. 1995;28:315–422. - PubMed
-
- Popot JL. Amphipols, nanodiscs, and fluorinated surfactants: three nonconventional approaches to studying membrane proteins in aqueous solutions. Annu Rev Biochem. 2010;79:737–75. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
