

Characterization of structural variants with single molecule and hybrid sequencing approaches
- PMID: 25355789
- PMCID: PMC4253835
- DOI: 10.1093/bioinformatics/btu714
Characterization of structural variants with single molecule and hybrid sequencing approaches
Abstract
Motivation: Structural variation is common in human and cancer genomes. High-throughput DNA sequencing has enabled genome-scale surveys of structural variation. However, the short reads produced by these technologies limit the study of complex variants, particularly those involving repetitive regions. Recent 'third-generation' sequencing technologies provide single-molecule templates and longer sequencing reads, but at the cost of higher per-nucleotide error rates.
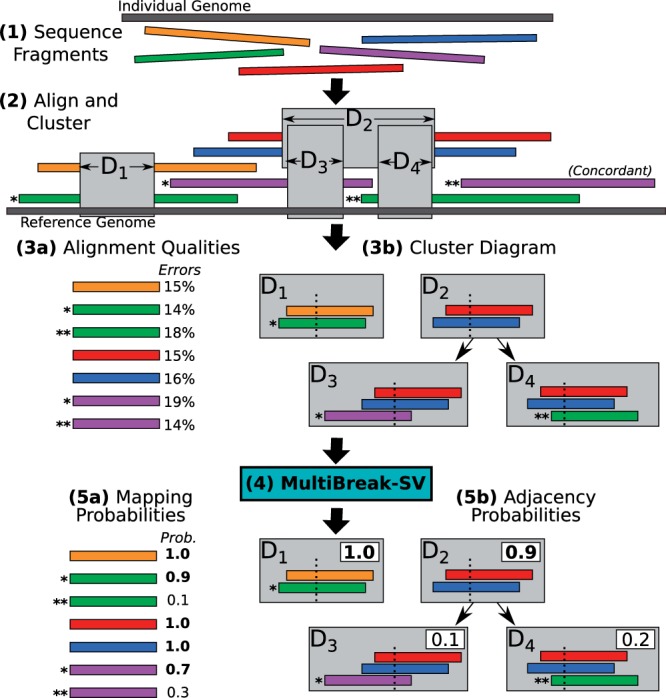
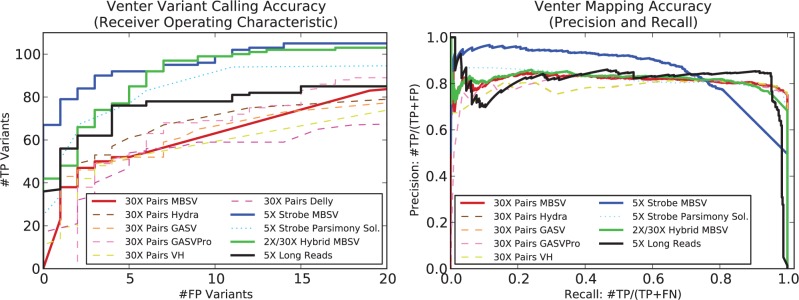
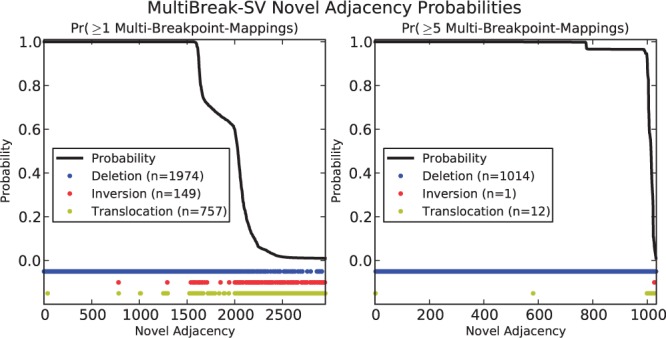
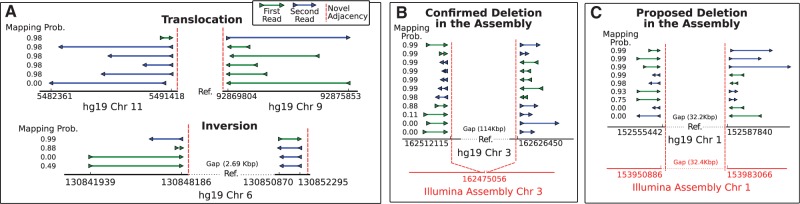
Results: We present MultiBreak-SV, an algorithm to detect structural variants (SVs) from single molecule sequencing data, paired read sequencing data, or a combination of sequencing data from different platforms. We demonstrate that combining low-coverage third-generation data from Pacific Biosciences (PacBio) with high-coverage paired read data is advantageous on simulated chromosomes. We apply MultiBreak-SV to PacBio data from four human fosmids and show that it detects known SVs with high sensitivity and specificity. Finally, we perform a whole-genome analysis on PacBio data from a complete hydatidiform mole cell line and predict 1002 high-probability SVs, over half of which are confirmed by an Illumina-based assembly.
© The Author 2014. Published by Oxford University Press. All rights reserved. For Permissions, please e-mail: journals.permissions@oup.com.
Figures
References
-
- Brown C. Single molecule strand sequencing using protein nanopores and scalable electronic devices. 2012 AGBT Conference.
