

Insight into the allosteric mechanism of Scapharca dimeric hemoglobin
- PMID: 25356908
- PMCID: PMC4245988
- DOI: 10.1021/bi500591s
Insight into the allosteric mechanism of Scapharca dimeric hemoglobin
Abstract
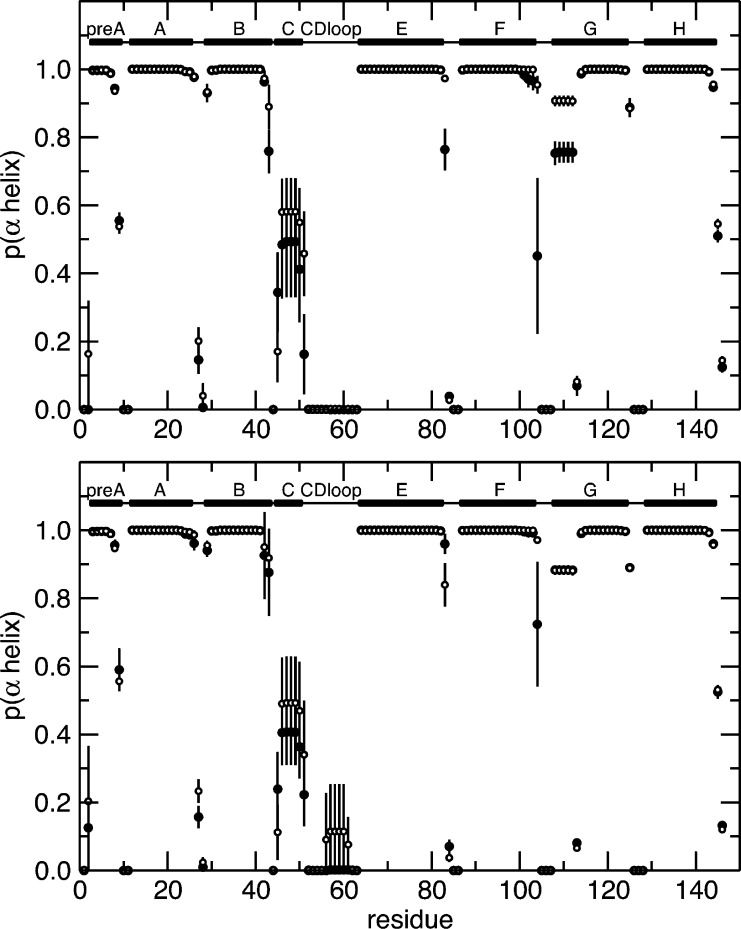
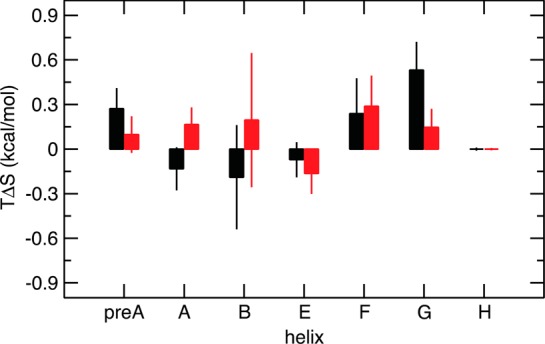
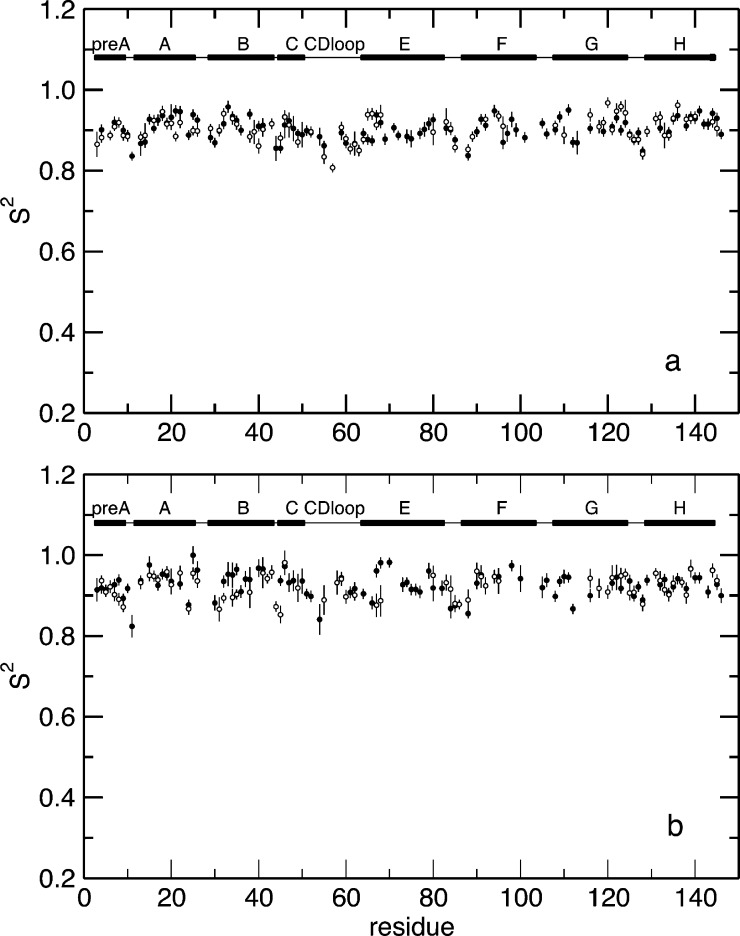
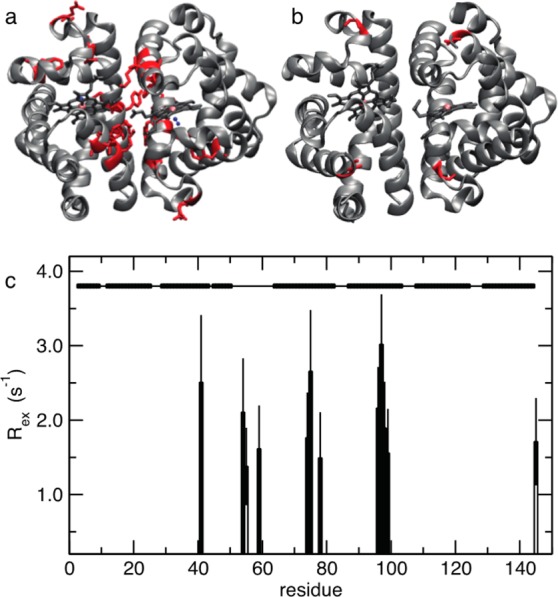
Allosteric regulation is an essential function of many proteins that control a variety of different processes such as catalysis, signal transduction, and gene regulation. Structural rearrangements have historically been considered the main means of communication between different parts of a protein. Recent studies have highlighted the importance, however, of changes in protein flexibility as an effective way to mediate allosteric communication across a protein. Scapharca dimeric hemoglobin (HbI) is the simplest possible allosteric system, with cooperative ligand binding between two identical subunits. Thermodynamic equilibrium studies of the binding of oxygen to HbI have shown that cooperativity is an entropically driven effect. The change in entropy of the system observed upon ligand binding may arise from changes in the protein, the ligand, or the water of the system. The goal of this study is to determine the contribution of the change in entropy of the protein backbone to HbI cooperative binding. Molecular dynamics simulations and nuclear magnetic resonance relaxation techniques have revealed that the fast internal motions of HbI contribute to the cooperative binding to carbon monoxide in two ways: (1) by contributing favorably to the free energy of the system and (2) by participating in the cooperative mechanism at the HbI subunit interface. The internal dynamics of the weakly cooperative HbI mutant, F97Y, were also investigated with the same methods. The changes in backbone NH dynamics observed for F97Y HbI upon ligand binding are not as large as for the wild type, in agreement with the reduced cooperativity observed for this mutant. The results of this study indicate that interface flexibility and backbone conformational entropy of HbI participate in and are important for the cooperative mechanism of carbon monoxide binding.
Figures
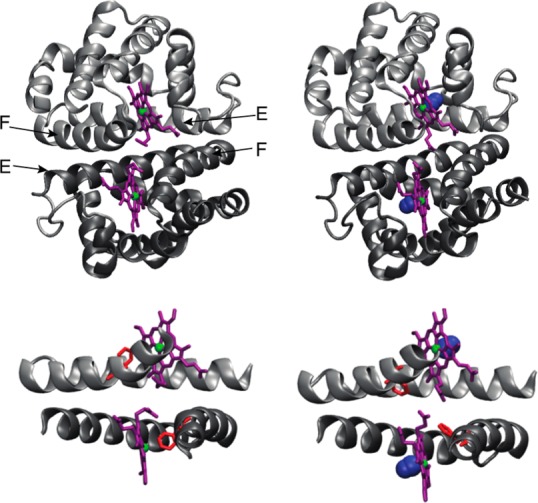
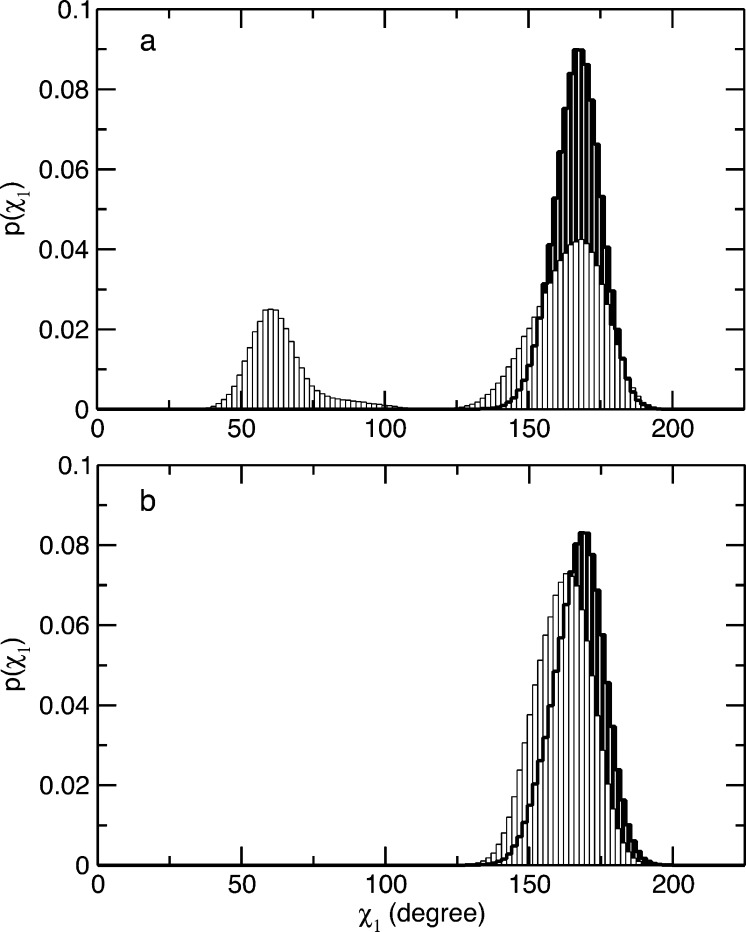
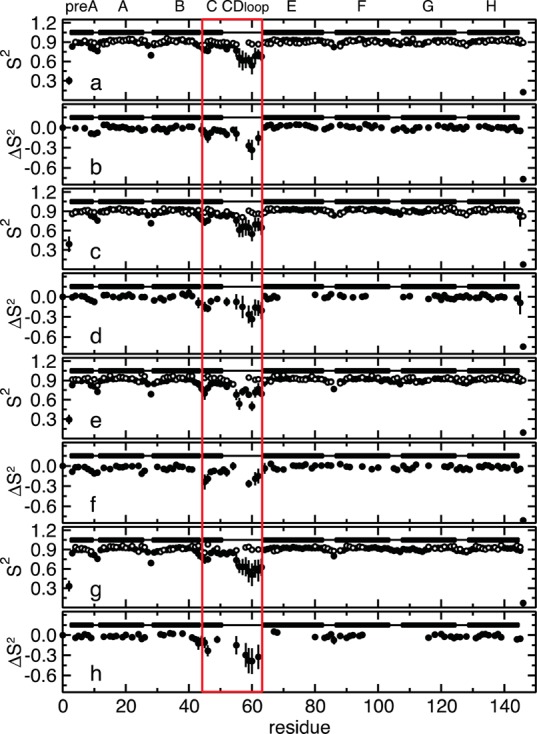
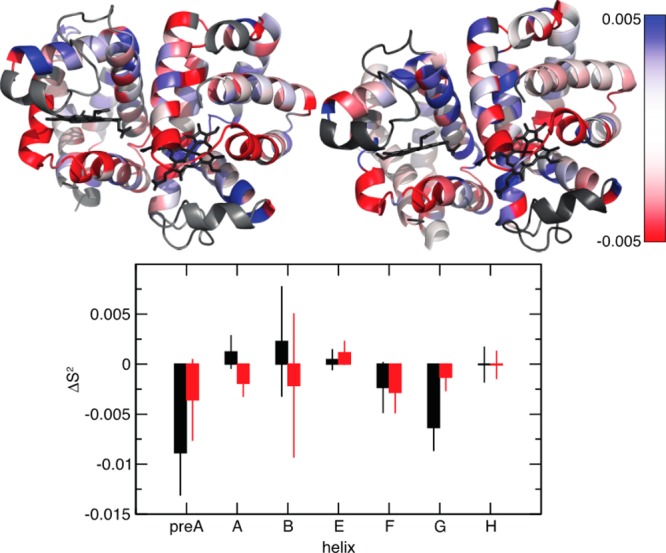

Similar articles
-
Protein Structural Dynamics of Wild-Type and Mutant Homodimeric Hemoglobin Studied by Time-Resolved X-Ray Solution Scattering.Int J Mol Sci. 2018 Nov 18;19(11):3633. doi: 10.3390/ijms19113633. Int J Mol Sci. 2018. PMID: 30453670 Free PMC article. Review.
-
Tertiary and quaternary allostery in tetrameric hemoglobin from Scapharca inaequivalvis.Biochemistry. 2013 Mar 26;52(12):2108-17. doi: 10.1021/bi301620x. Epub 2013 Mar 15. Biochemistry. 2013. PMID: 23458680 Free PMC article.
-
Cooperative Protein Dynamics of Heterotetrameric Hemoglobin from Scapharca inaequivalvis.J Phys Chem B. 2024 Aug 8;128(31):7558-7567. doi: 10.1021/acs.jpcb.4c03917. Epub 2024 Jul 29. J Phys Chem B. 2024. PMID: 39072557
-
Interfacial water effect on cooperativity and signal communication in Scapharca dimeric hemoglobin.Phys Chem Chem Phys. 2017 Mar 8;19(10):7380-7389. doi: 10.1039/c7cp00280g. Phys Chem Chem Phys. 2017. PMID: 28243652
-
Structural and thermodynamic aspects of cooperativity in the homodimeric hemoglobin from Scapharca inaequivalvis.Biophys Chem. 2000 Aug 30;86(2-3):173-8. doi: 10.1016/s0301-4622(00)00162-9. Biophys Chem. 2000. PMID: 11026682 Review.
Cited by
-
Ligand and interfacial dynamics in a homodimeric hemoglobin.Struct Dyn. 2016 Feb 17;3(1):012003. doi: 10.1063/1.4940228. eCollection 2016 Jan. Struct Dyn. 2016. PMID: 26958581 Free PMC article.
-
Genetic Mutations in the S-loop of Human Glutathione Synthetase: Links Between Substrate Binding, Active Site Structure and Allostery.Comput Struct Biotechnol J. 2018 Nov 29;17:31-38. doi: 10.1016/j.csbj.2018.11.008. eCollection 2019. Comput Struct Biotechnol J. 2018. PMID: 30581542 Free PMC article.
-
Effects of mutations on the molecular dynamics of oxygen escape from the dimeric hemoglobin of Scapharca inaequivalvis.F1000Res. 2015 Mar 13;4:65. doi: 10.12688/f1000research.6127.1. eCollection 2015. F1000Res. 2015. PMID: 25866622 Free PMC article.
-
Protein Structural Dynamics of Wild-Type and Mutant Homodimeric Hemoglobin Studied by Time-Resolved X-Ray Solution Scattering.Int J Mol Sci. 2018 Nov 18;19(11):3633. doi: 10.3390/ijms19113633. Int J Mol Sci. 2018. PMID: 30453670 Free PMC article. Review.
-
Conformational Dynamics of Phytoglobin BvPgb1.2 from Beta vulgaris ssp. vulgaris.Int J Mol Sci. 2023 Feb 16;24(4):3973. doi: 10.3390/ijms24043973. Int J Mol Sci. 2023. PMID: 36835381 Free PMC article.
References
-
- Pardee A. B.; Reddy G. P. (2003) Beginnings of feedback inhibition, allostery, and multi-protein complexes. Gene 321, 17–23. - PubMed
-
- Fuxe K.; Borroto-Escuela D. O.; Marcellino D.; Romero-Fernandez W.; Frankowska M.; Guidolin D.; Filip M.; Ferraro L.; Woods A. S.; Tarakanov A.; Ciruela F.; Agnati L. F.; Tanganelli S. (2012) GPCR heteromers and their allosteric receptor-receptor interactions. Curr. Med. Chem. 19, 356–363. - PubMed
-
- Kalodimos C. G. (2012) Protein function and allostery: A dynamic relationship. Ann. N.Y. Acad. Sci. 1260, 81–86. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
