

Genome-wide distribution of RNA-DNA hybrids identifies RNase H targets in tRNA genes, retrotransposons and mitochondria
- PMID: 25357144
- PMCID: PMC4214602
- DOI: 10.1371/journal.pgen.1004716
Genome-wide distribution of RNA-DNA hybrids identifies RNase H targets in tRNA genes, retrotransposons and mitochondria
Abstract
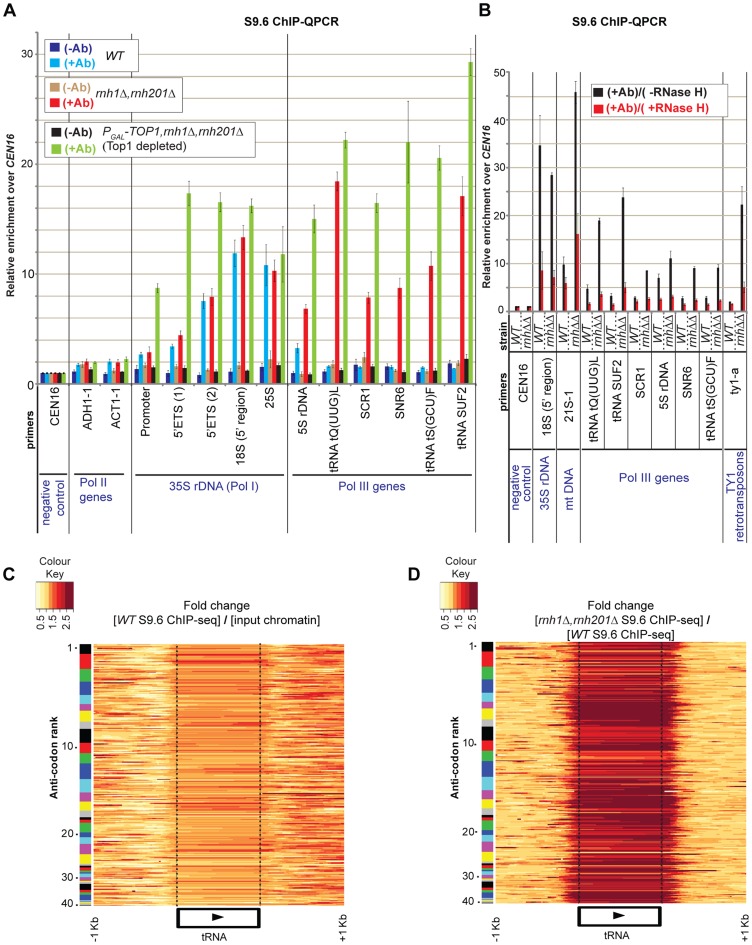
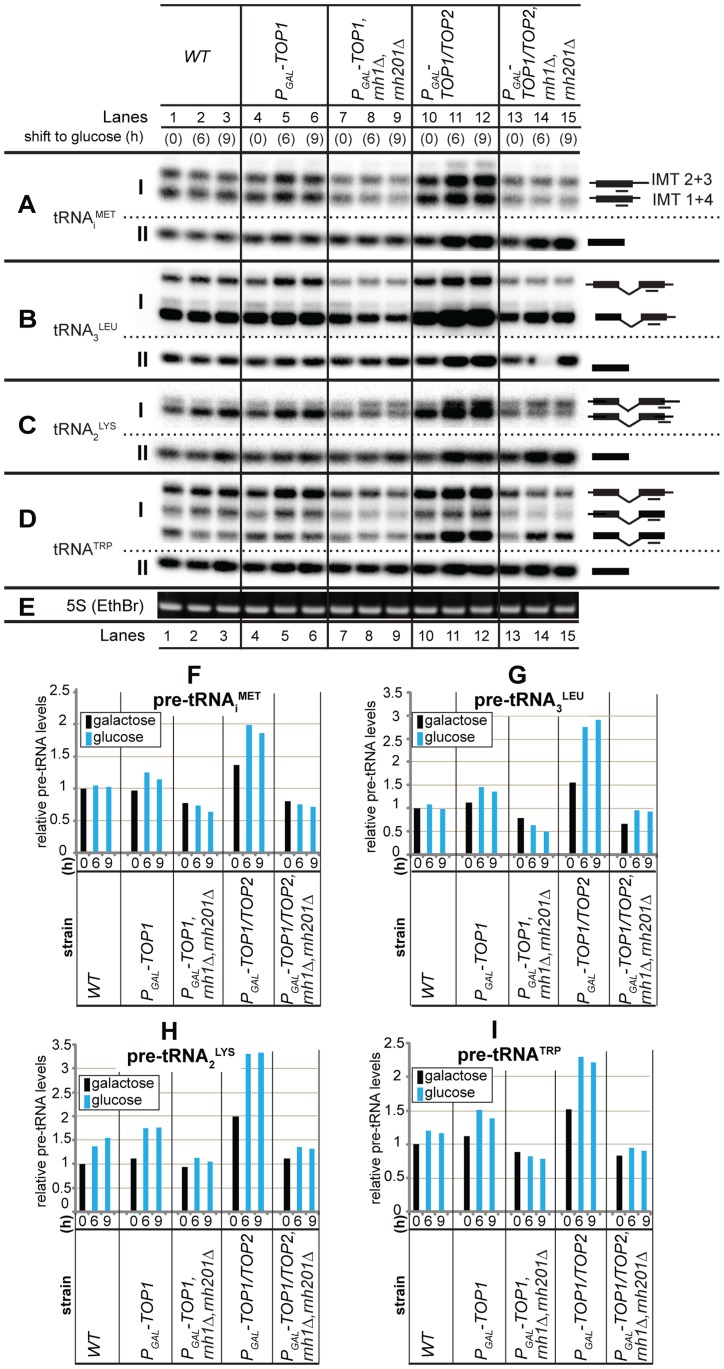
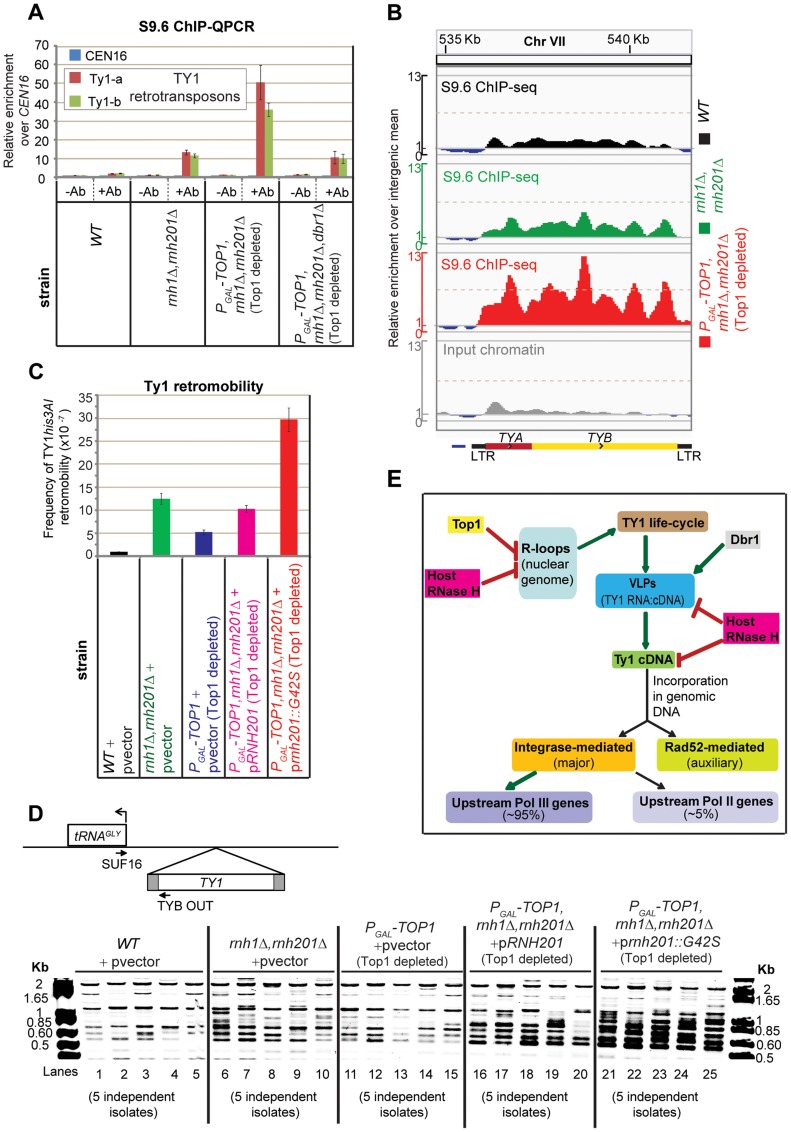
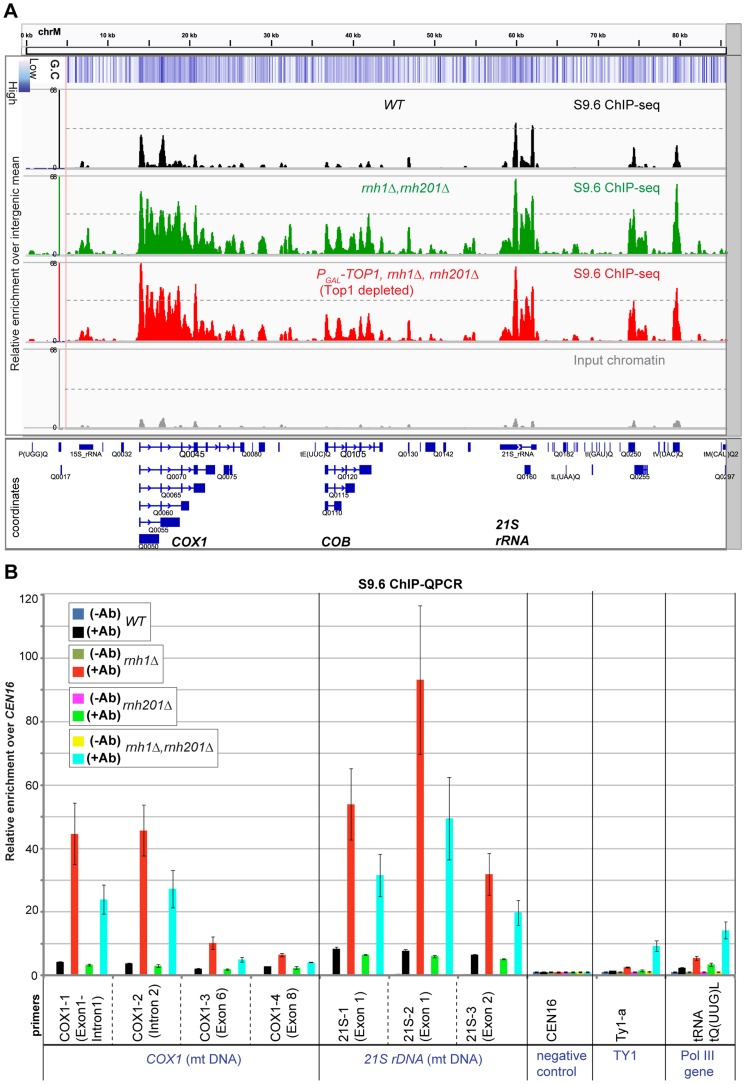
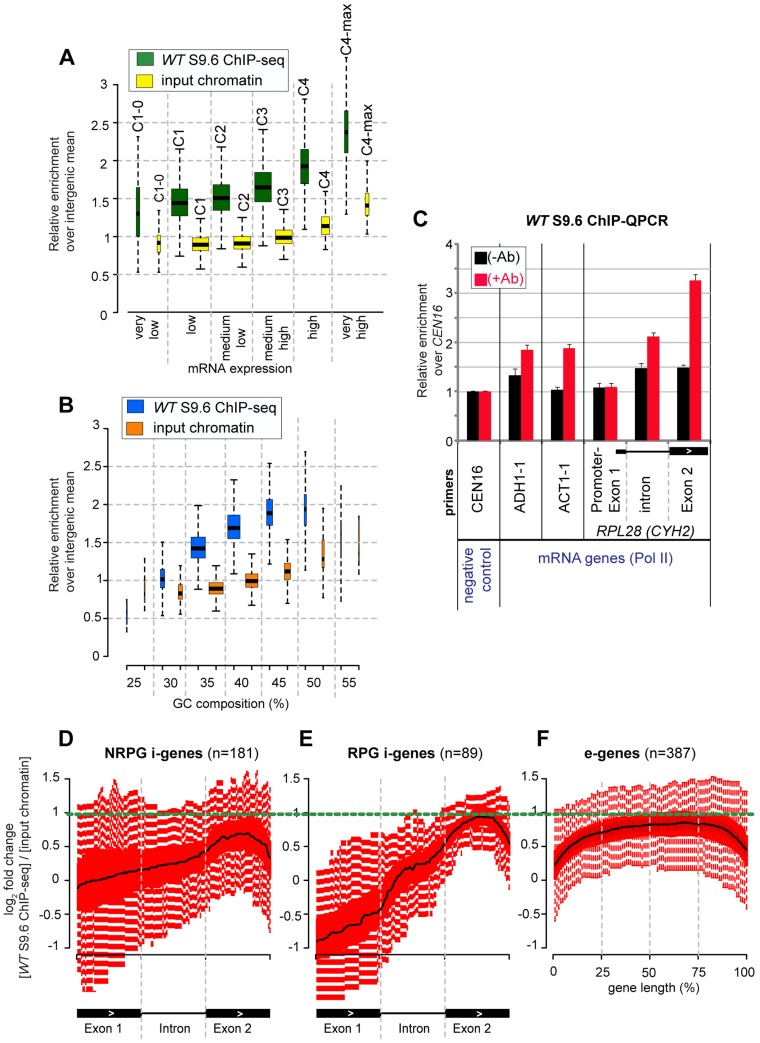
During transcription, the nascent RNA can invade the DNA template, forming extended RNA-DNA duplexes (R-loops). Here we employ ChIP-seq in strains expressing or lacking RNase H to map targets of RNase H activity throughout the budding yeast genome. In wild-type strains, R-loops were readily detected over the 35S rDNA region, transcribed by Pol I, and over the 5S rDNA, transcribed by Pol III. In strains lacking RNase H activity, R-loops were elevated over other Pol III genes, notably tRNAs, SCR1 and U6 snRNA, and were also associated with the cDNAs of endogenous TY1 retrotransposons, which showed increased rates of mobility to the 5'-flanking regions of tRNA genes. Unexpectedly, R-loops were also associated with mitochondrial genes in the absence of RNase H1, but not of RNase H2. Finally, R-loops were detected on actively transcribed protein-coding genes in the wild-type, particularly over the second exon of spliced ribosomal protein genes.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Aguilera A, Garcia-Muse T (2012) R loops: from transcription byproducts to threats to genome stability. Mol Cell 46: 115–124. - PubMed
-
- Belotserkovskii BP, Mirkin SM, Hanawalt PC (2013) DNA Sequences That Interfere with Transcription: Implications for Genome Function and Stability. Chem Rev 113: 8620–8637. - PubMed
-
- Drolet M (2006) Growth inhibition mediated by excess negative supercoiling: the interplay between transcription elongation, R-loop formation and DNA topology. Mol Microbiol 59: 723–730. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
