

Proteogenomics: concepts, applications and computational strategies
- PMID: 25357241
- PMCID: PMC4392723
- DOI: 10.1038/nmeth.3144
Proteogenomics: concepts, applications and computational strategies
Abstract
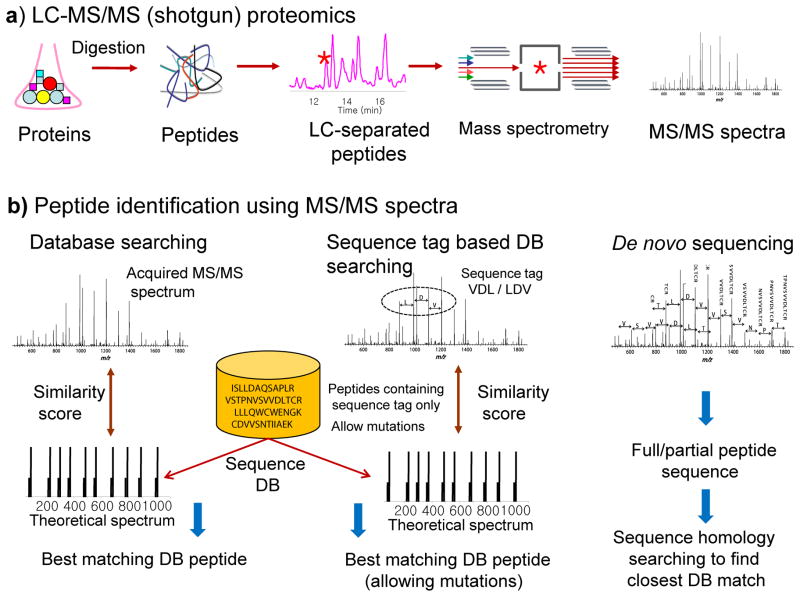
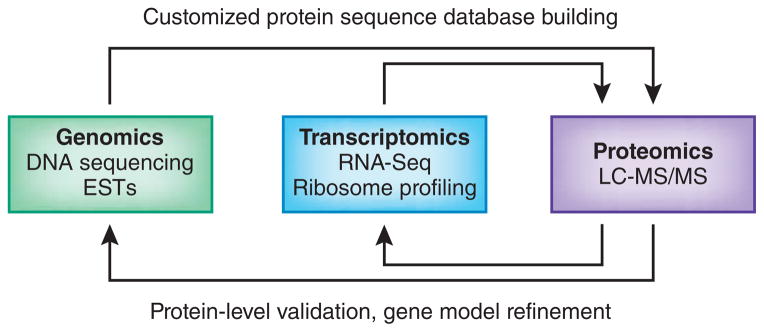
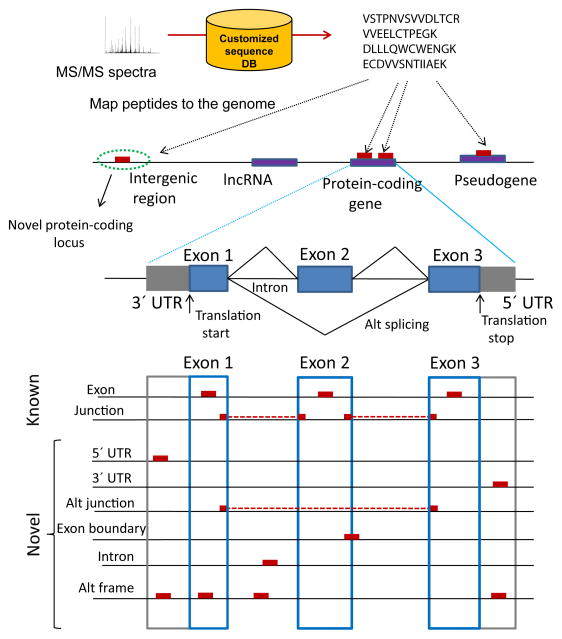
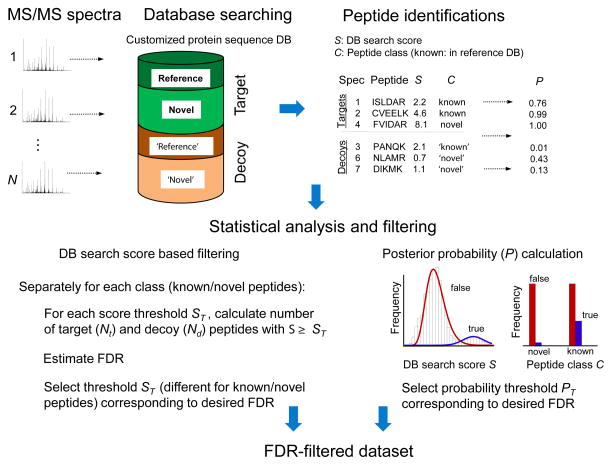
Proteogenomics is an area of research at the interface of proteomics and genomics. In this approach, customized protein sequence databases generated using genomic and transcriptomic information are used to help identify novel peptides (not present in reference protein sequence databases) from mass spectrometry-based proteomic data; in turn, the proteomic data can be used to provide protein-level evidence of gene expression and to help refine gene models. In recent years, owing to the emergence of new sequencing technologies such as RNA-seq and dramatic improvements in the depth and throughput of mass spectrometry-based proteomics, the pace of proteogenomic research has greatly accelerated. Here I review the current state of proteogenomic methods and applications, including computational strategies for building and using customized protein sequence databases. I also draw attention to the challenge of false positive identifications in proteogenomics and provide guidelines for analyzing the data and reporting the results of proteogenomic studies.
Conflict of interest statement
The author declares no competing financial interests.
Figures
References
-
- Mann M, Kulak NA, Nagaraj N, Cox J. The coming age of complete, accurate, and ubiquitous proteomes. Mol Cell. 2013;49:583–590. - PubMed
-
- Bantscheff M, Lemeer S, Savitski MM, Kuster B. Quantitative mass spectrometry in proteomics: critical review update from 2007 to the present. Anal Bioanal Chem. 2012;404:939–965. - PubMed
-
- Nesvizhskii AI, Aebersold R. Interpretation of shotgun proteomic data - The protein inference problem. Molecular & Cellular Proteomics. 2005;4:1419–1440. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
