

Preclinical efficacy of MEK inhibition in Nras-mutant AML
- PMID: 25361812
- PMCID: PMC4271180
- DOI: 10.1182/blood-2014-05-574582
Preclinical efficacy of MEK inhibition in Nras-mutant AML
Abstract
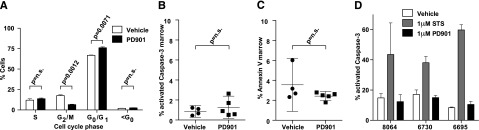
Oncogenic NRAS mutations are highly prevalent in acute myeloid leukemia (AML). Genetic analysis supports the hypothesis that NRAS mutations cooperate with antecedent molecular lesions in leukemogenesis, but have limited independent prognostic significance. Using short hairpin RNA-mediated knockdown in human cell lines and primary mouse leukemias, we show that AML cells with NRAS/Nras mutations are dependent on continued oncogene expression in vitro and in vivo. Using the Mx1-Cre transgene to inactivate a conditional mutant Nras allele, we analyzed hematopoiesis and hematopoietic stem and progenitor cells (HSPCs) under normal and stressed conditions and found that HSPCs lacking Nras expression are functionally equivalent to normal HSPCs in the adult mouse. Treating recipient mice transplanted with primary Nras(G12D) AMLs with 2 potent allosteric mitogen-activated protein kinase kinase (MEK) inhibitors (PD0325901 or trametinib/GlaxoSmithKline 1120212) significantly prolonged survival and reduced proliferation but did not induce apoptosis, promote differentiation, or drive clonal evolution. The phosphatidylinositol 3-kinase inhibitor GDC-0941 was ineffective as a single agent and did not augment the activity of PD0325901. All mice ultimately succumbed to progressive leukemia. Together, these data validate oncogenic N-Ras signaling as a therapeutic target in AML and support testing combination regimens that include MEK inhibitors.
© 2014 by The American Society of Hematology.
Figures
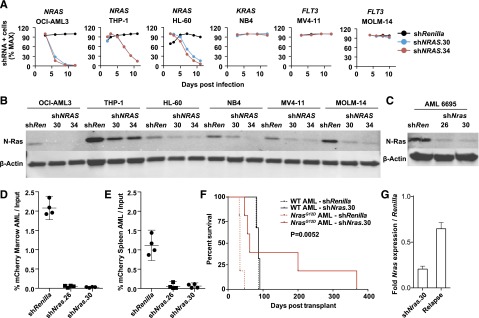
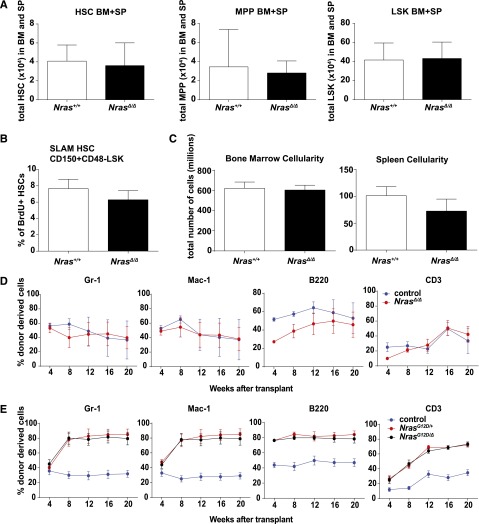
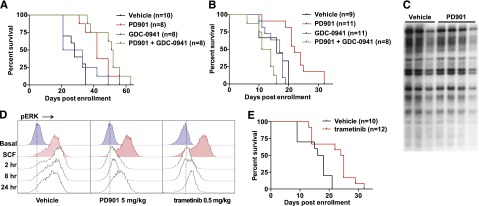
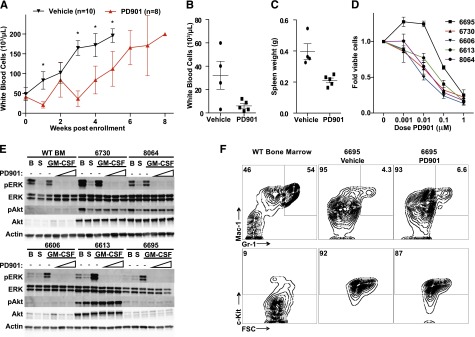
References
-
- Bachas C, Schuurhuis GJ, Hollink IH, et al. High-frequency type I/II mutational shifts between diagnosis and relapse are associated with outcome in pediatric AML: implications for personalized medicine. Blood. 2010;116(15):2752–2758. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
