

Evaluation of variant identification methods for whole genome sequencing data in dairy cattle
- PMID: 25361890
- PMCID: PMC4289218
- DOI: 10.1186/1471-2164-15-948
Evaluation of variant identification methods for whole genome sequencing data in dairy cattle
Abstract
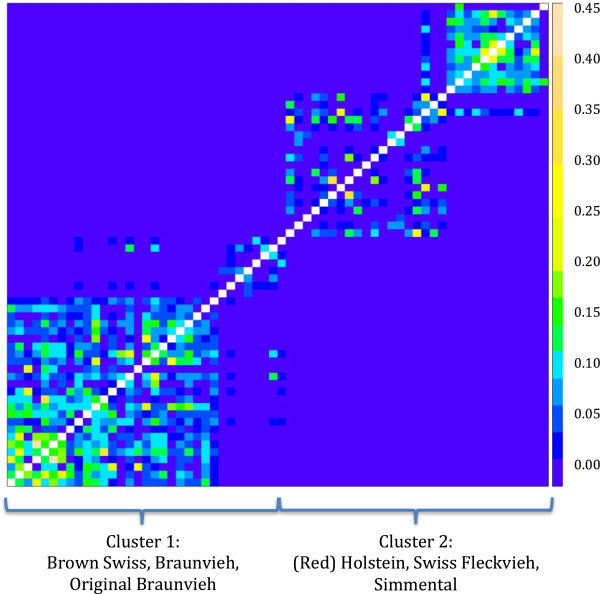
Background: Advances in human genomics have allowed unprecedented productivity in terms of algorithms, software, and literature available for translating raw next-generation sequence data into high-quality information. The challenges of variant identification in organisms with lower quality reference genomes are less well documented. We explored the consequences of commonly recommended preparatory steps and the effects of single and multi sample variant identification methods using four publicly available software applications (Platypus, HaplotypeCaller, Samtools and UnifiedGenotyper) on whole genome sequence data of 65 key ancestors of Swiss dairy cattle populations. Accuracy of calling next-generation sequence variants was assessed by comparison to the same loci from medium and high-density single nucleotide variant (SNV) arrays.
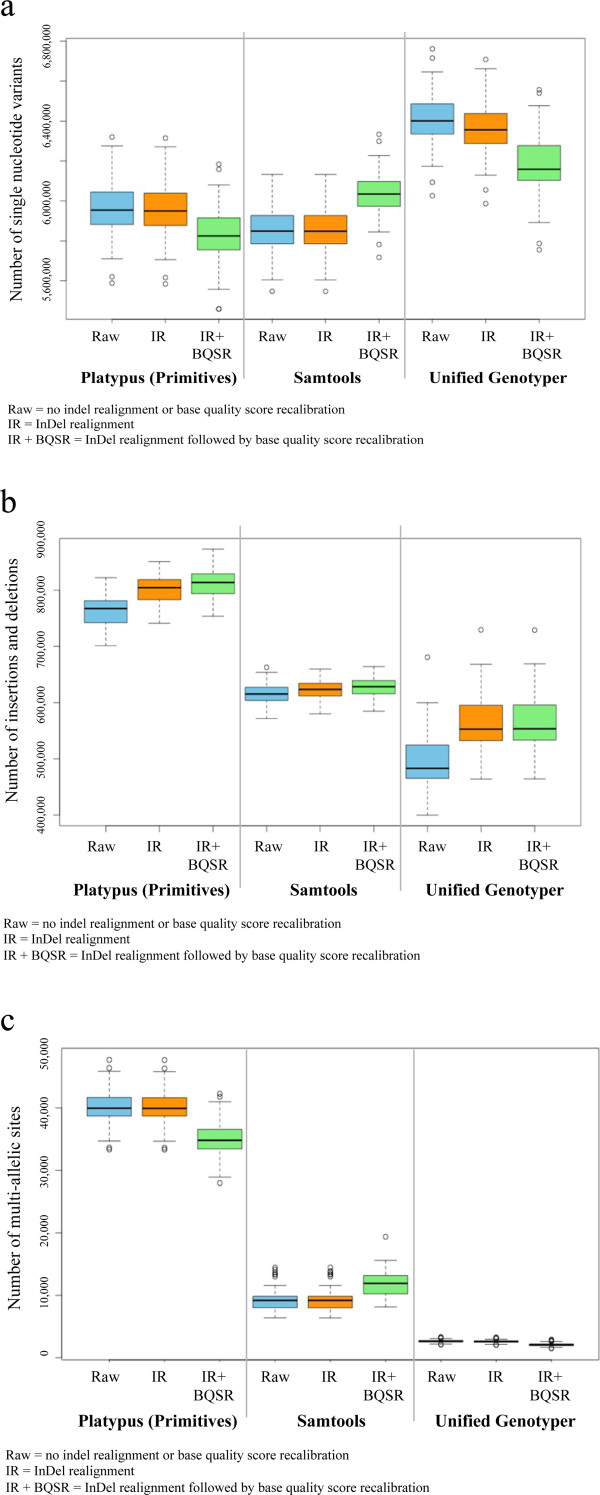
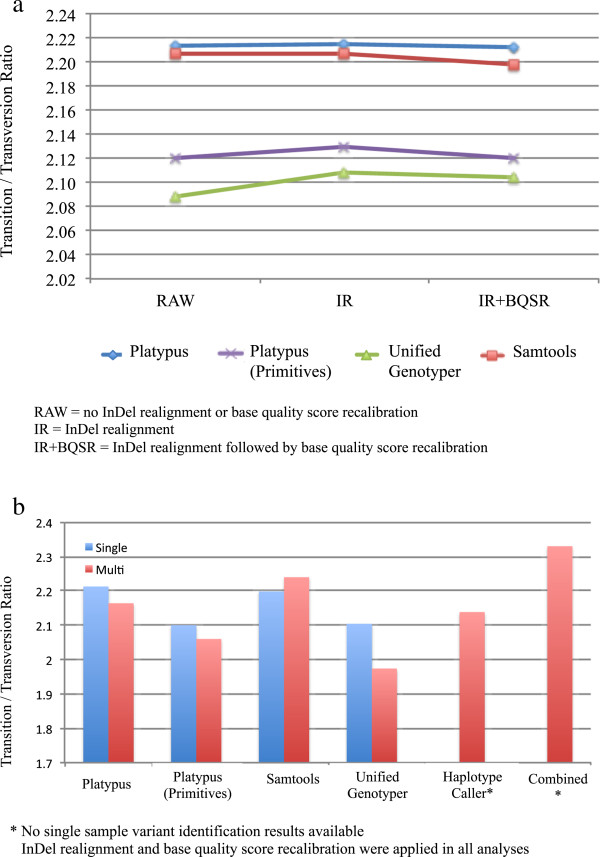
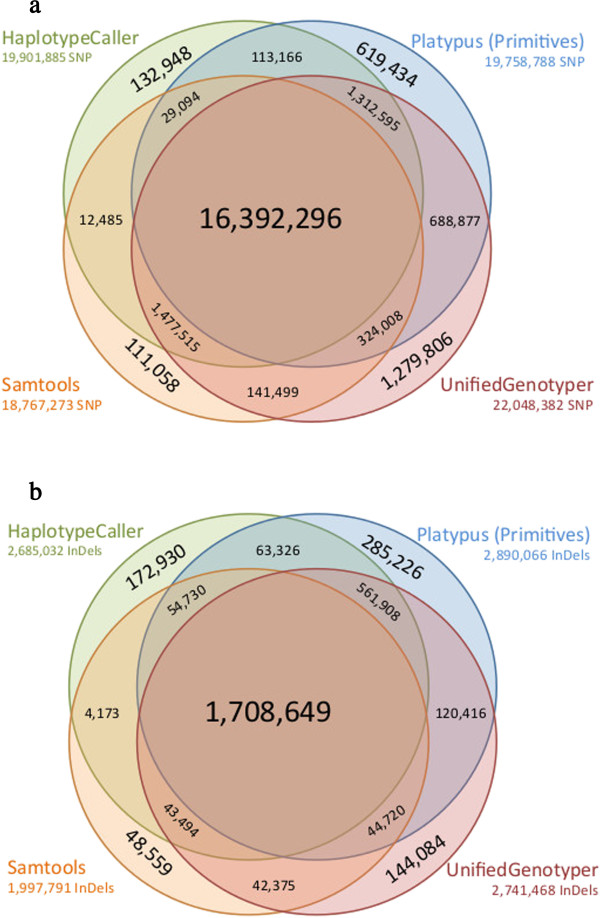
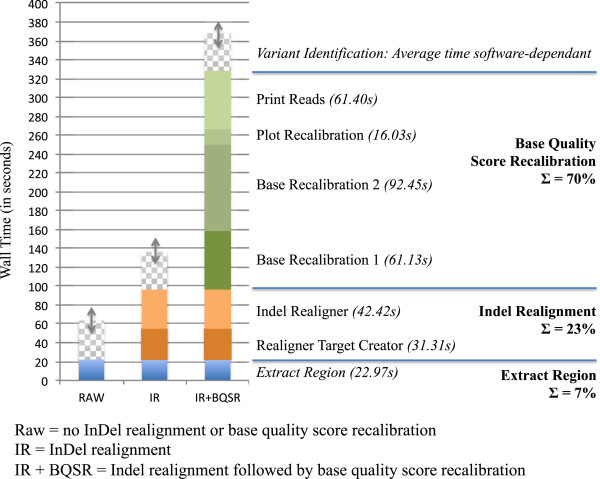
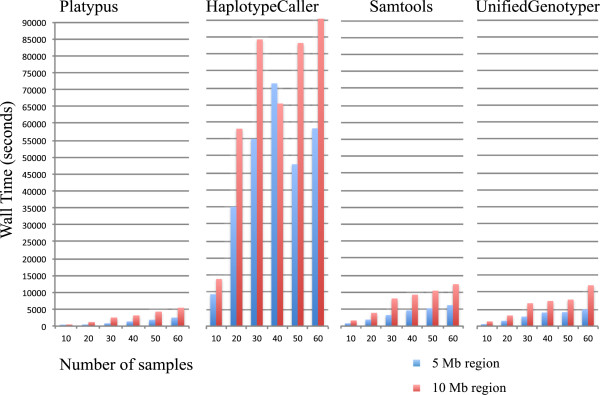
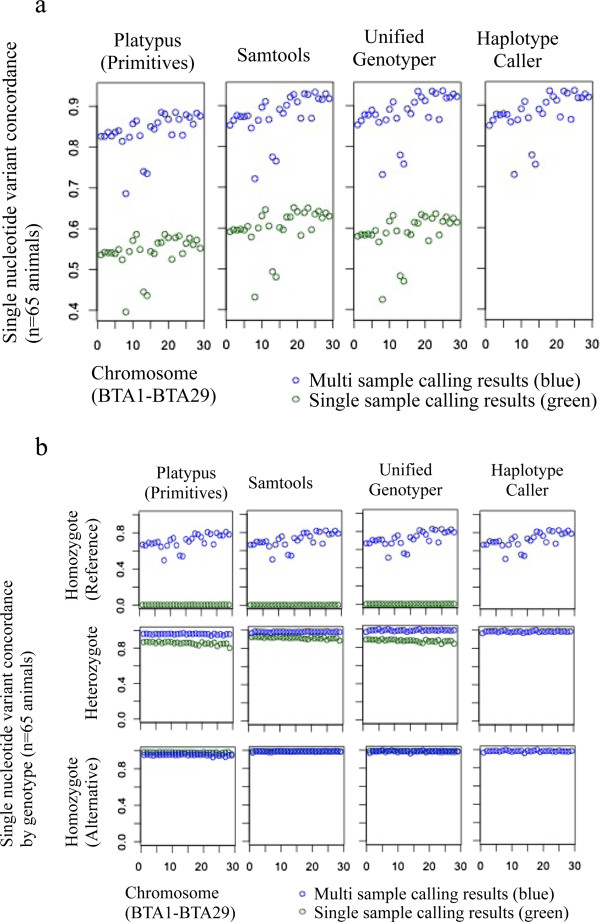
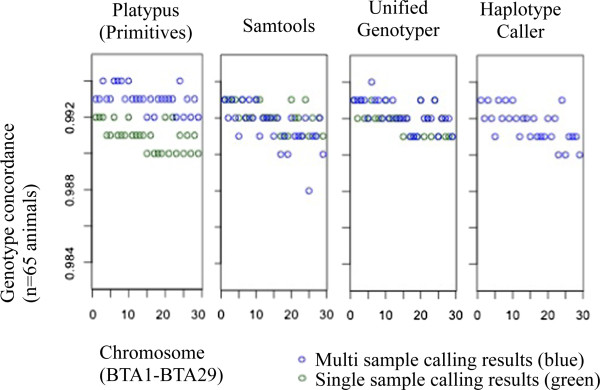
Results: The total number of SNVs identified varied by software and method, with single (multi) sample results ranging from 17.7 to 22.0 (16.9 to 22.0) million variants. Computing time varied considerably between software. Preparatory realignment of insertions and deletions and subsequent base quality score recalibration had only minor effects on the number and quality of SNVs identified by different software, but increased computing time considerably. Average concordance for single (multi) sample results with high-density chip data was 58.3% (87.0%) and average genotype concordance in correctly identified SNVs was 99.2% (99.2%) across software. The average quality of SNVs identified, measured as the ratio of transitions to transversions, was higher using single sample methods than multi sample methods. A consensus approach using results of different software generally provided the highest variant quality in terms of transition/transversion ratio.
Conclusions: Our findings serve as a reference for variant identification pipeline development in non-human organisms and help assess the implication of preparatory steps in next-generation sequencing pipelines for organisms with incomplete reference genomes (pipeline code is included). Benchmarking this information should prove particularly useful in processing next-generation sequencing data for use in genome-wide association studies and genomic selection.
Figures


References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
