

A multiscale statistical mechanical framework integrates biophysical and genomic data to assemble cancer networks
- PMID: 25362484
- PMCID: PMC4244270
- DOI: 10.1038/ng.3138
A multiscale statistical mechanical framework integrates biophysical and genomic data to assemble cancer networks
Abstract
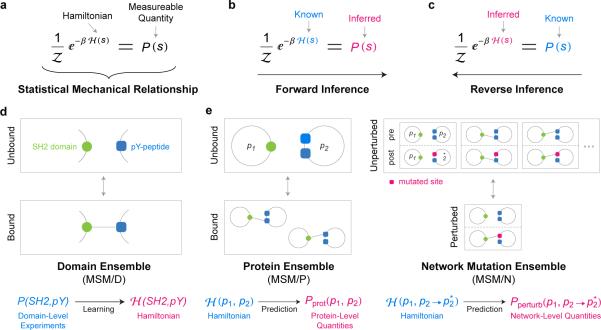
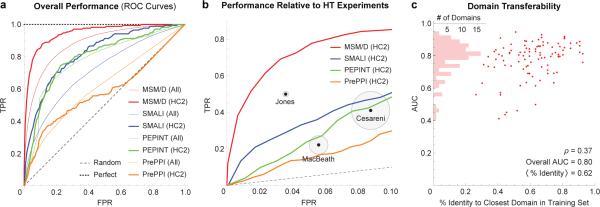
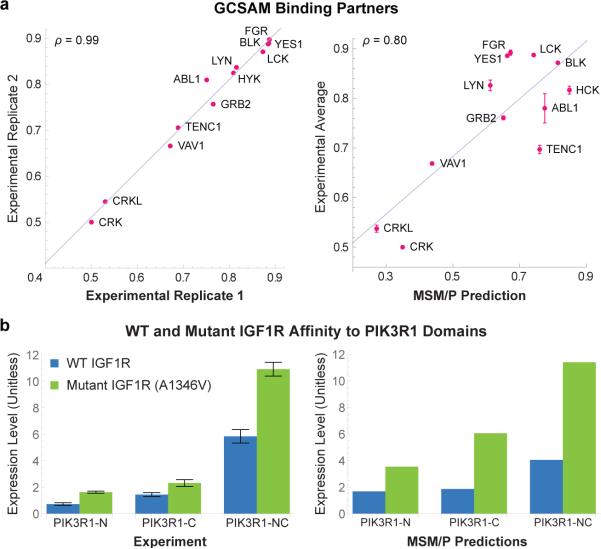
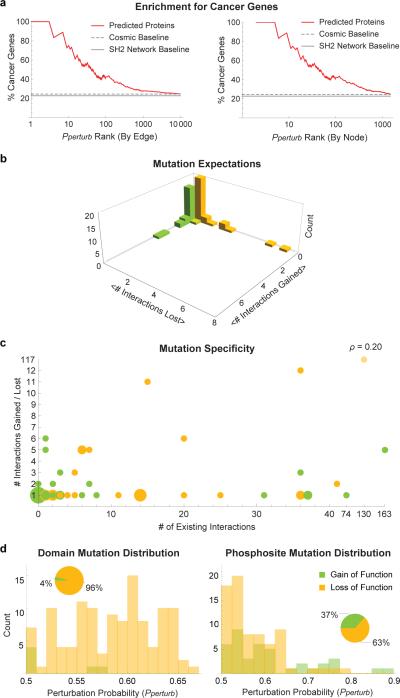
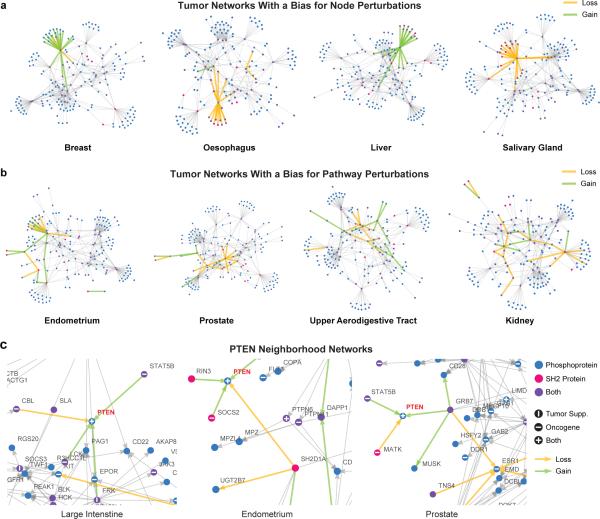
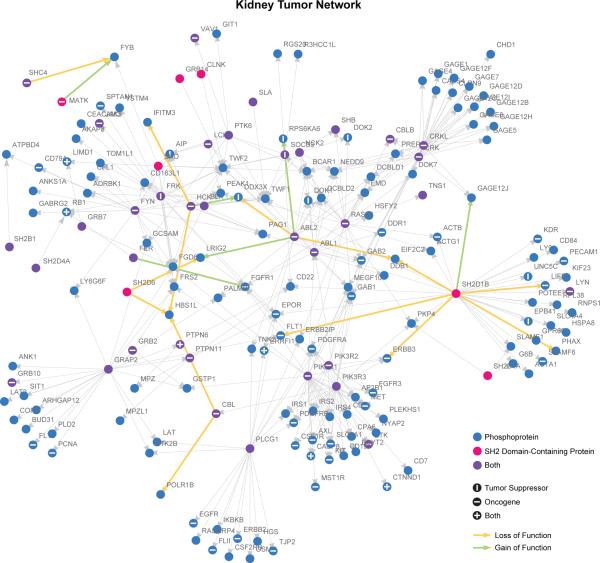
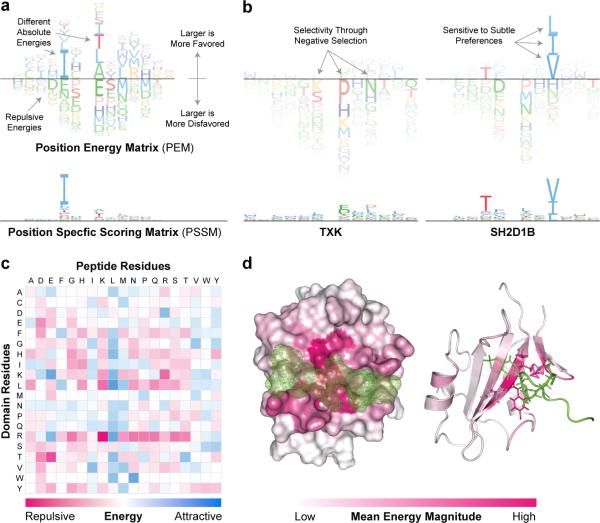
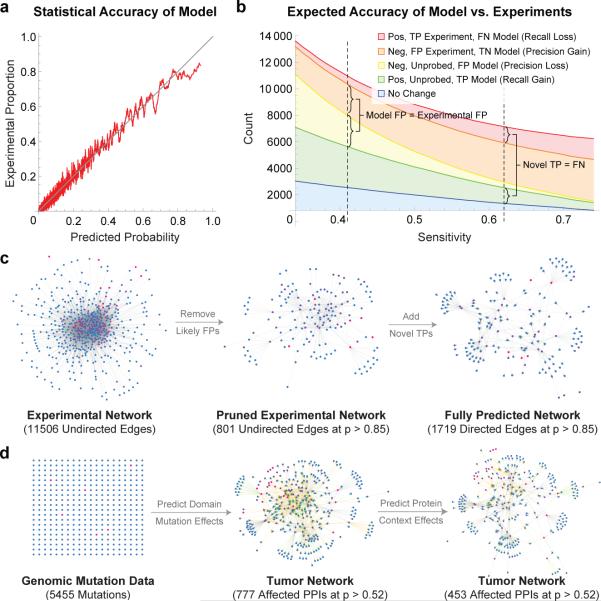
Functional interpretation of genomic variation is critical to understanding human disease, but it remains difficult to predict the effects of specific mutations on protein interaction networks and the phenotypes they regulate. We describe an analytical framework based on multiscale statistical mechanics that integrates genomic and biophysical data to model the human SH2-phosphoprotein network in normal and cancer cells. We apply our approach to data in The Cancer Genome Atlas (TCGA) and test model predictions experimentally. We find that mutations mapping to phosphoproteins often create new interactions but that mutations altering SH2 domains result almost exclusively in loss of interactions. Some of these mutations eliminate all interactions, but many cause more selective loss, thereby rewiring specific edges in highly connected subnetworks. Moreover, idiosyncratic mutations appear to be as functionally consequential as recurrent mutations. By synthesizing genomic, structural and biochemical data, our framework represents a new approach to the interpretation of genetic variation.
Figures
Comment in
-
Predicting protein networks in cancer.Nat Genet. 2014 Dec;46(12):1252-3. doi: 10.1038/ng.3156. Nat Genet. 2014. PMID: 25418743
References
-
- Ho Y, et al. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature. 2002;415:180–183. - PubMed
-
- Bader GD, Hogue CWV. Analyzing yeast protein–protein interaction data obtained from different sources. Nat. Biotechnol. 2002;2020:991, 991–997. - PubMed
-
- Gschwind A, Fischer OM, Ullrich A. The discovery of receptor tyrosine kinases: targets for cancer therapy. Nat. Rev. Cancer. 2004;4:361–370. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
