

Cellular prion protein and NMDA receptor modulation: protecting against excitotoxicity
- PMID: 25364752
- PMCID: PMC4207032
- DOI: 10.3389/fcell.2014.00045
Cellular prion protein and NMDA receptor modulation: protecting against excitotoxicity
Abstract
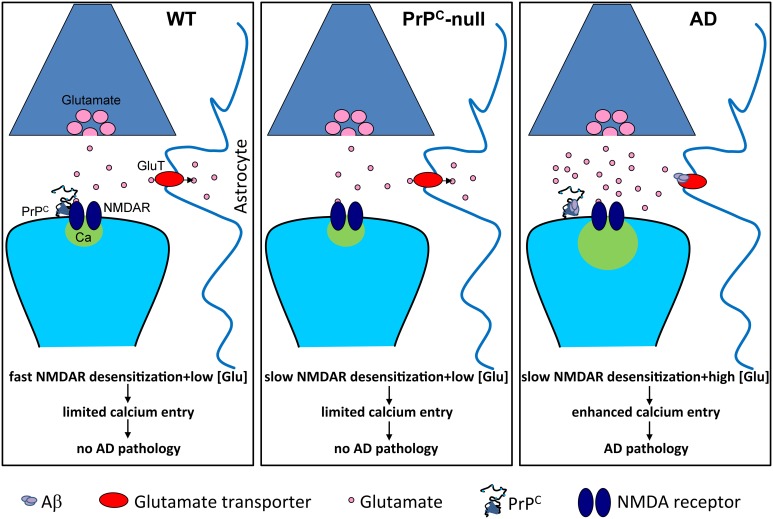
Although it is well established that misfolding of the cellular prion protein (PrP(C)) into the β-sheet-rich, aggregated scrapie conformation (PrP(Sc)) causes a variety of transmissible spongiform encephalopathies (TSEs), the physiological roles of PrP(C) are still incompletely understood. There is accumulating evidence describing the roles of PrP(C) in neurodegeneration and neuroinflammation. Recently, we identified a functional regulation of NMDA receptors by PrP(C) that involves formation of a physical protein complex between these proteins. Excessive NMDA receptor activity during conditions such as ischemia mediates enhanced Ca(2+) entry into cells and contributes to excitotoxic neuronal death. In addition, NMDA receptors and/or PrP(C) play critical roles in neuroinflammation and glial cell toxicity. Inhibition of NMDA receptor activity protects against PrP(Sc)-induced neuronal death. Moreover, in mice lacking PrP(C), infarct size is increased after focal cerebral ischemia, and absence of PrP(C) increases susceptibility of neurons to NMDA receptor-dependent death. Recently, PrP(C) was found to be a receptor for oligomeric beta-amyloid (Aβ) peptides, suggesting a role for PrP(C) in Alzheimer's disease (AD). Our recent findings suggest that Aβ peptides enhance NMDA receptor current by perturbing the normal copper- and PrP(C)-dependent regulation of these receptors. Here, we review evidence highlighting a role for PrP(C) in preventing NMDA receptor-mediated excitotoxicity and inflammation. There is a need for more detailed molecular characterization of PrP(C)-mediated regulation of NMDA receptors, such as determining which NMDA receptor subunits mediate pathogenic effects upon loss of PrP(C)-mediated regulation and identifying PrP(C) binding site(s) on the receptor. This knowledge will allow development of novel therapeutic interventions for not only TSEs, but also for AD and other neurodegenerative disorders involving dysfunction of PrP(C).
Keywords: Alzheimer's disease; NMDA receptor; beta-amyloid; cellular prion protein; excitotoxicity; ischemia; neuroinflammation.
Figures

References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous