

A census of human RNA-binding proteins
- PMID: 25365966
- PMCID: PMC11148870
- DOI: 10.1038/nrg3813
A census of human RNA-binding proteins
Abstract
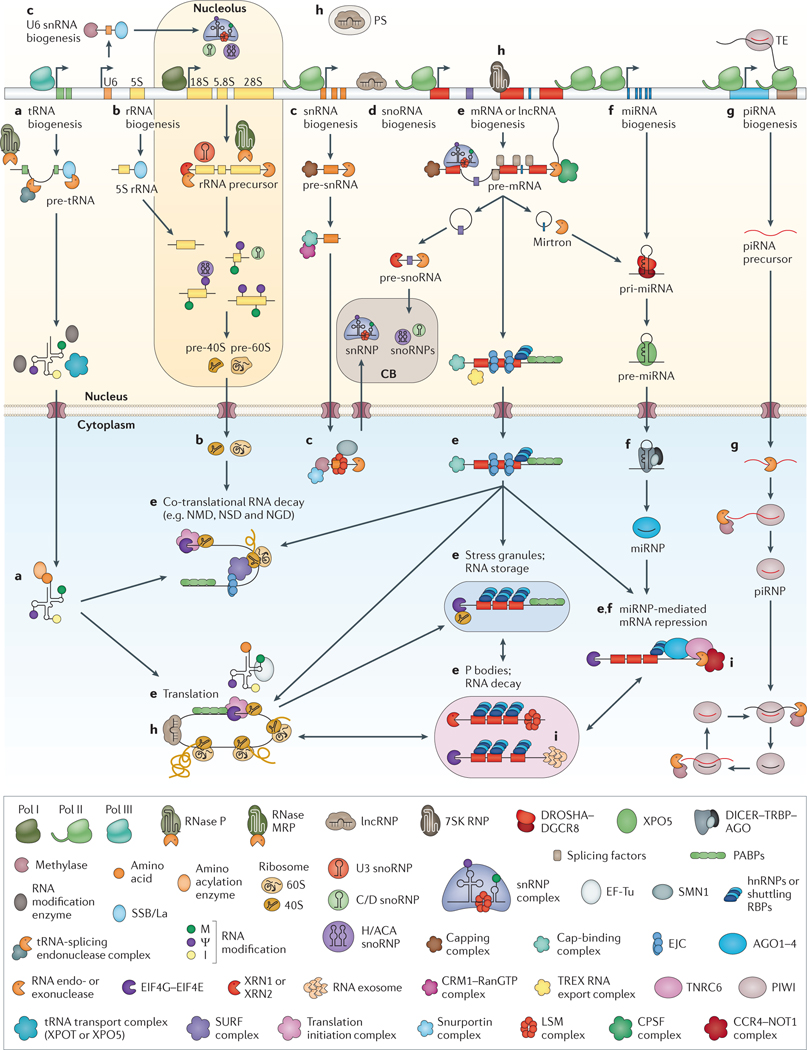
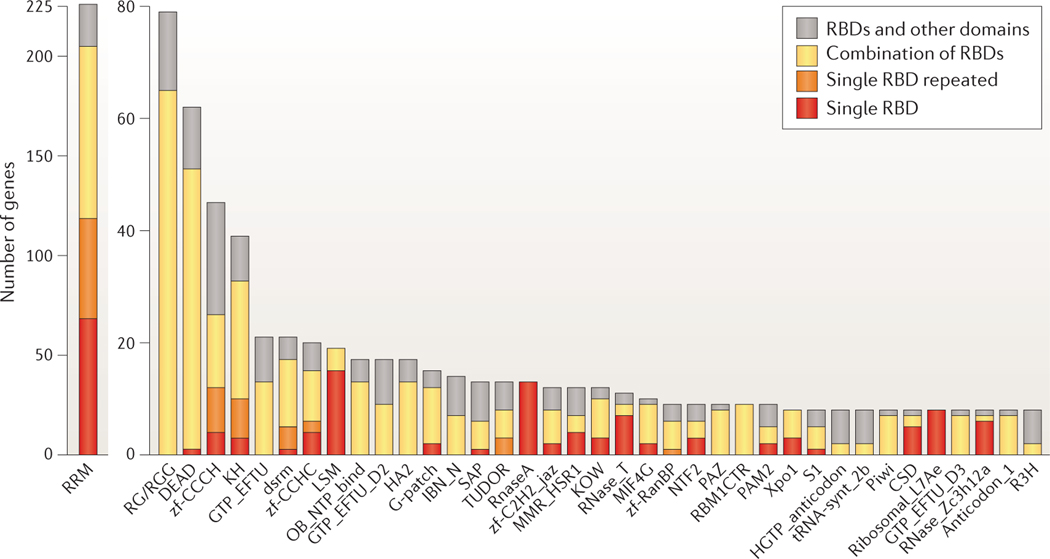
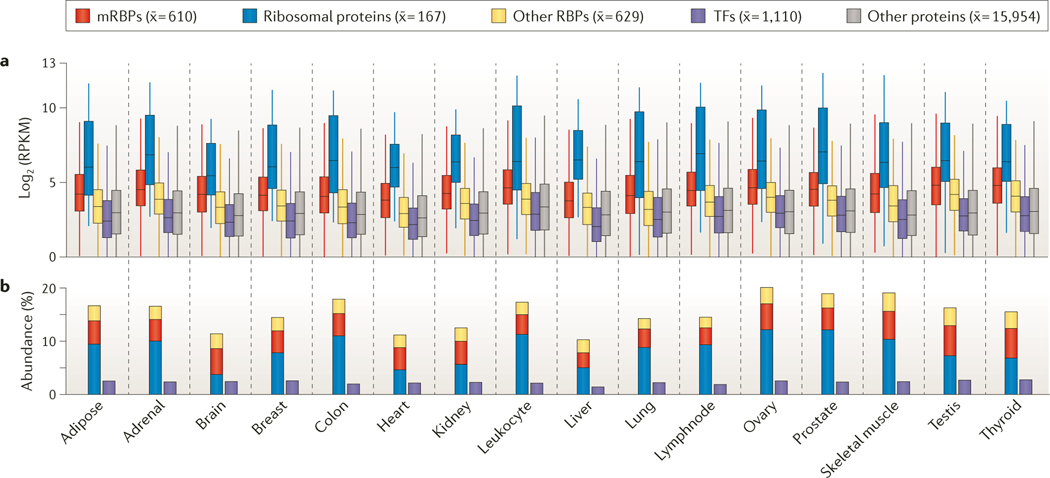
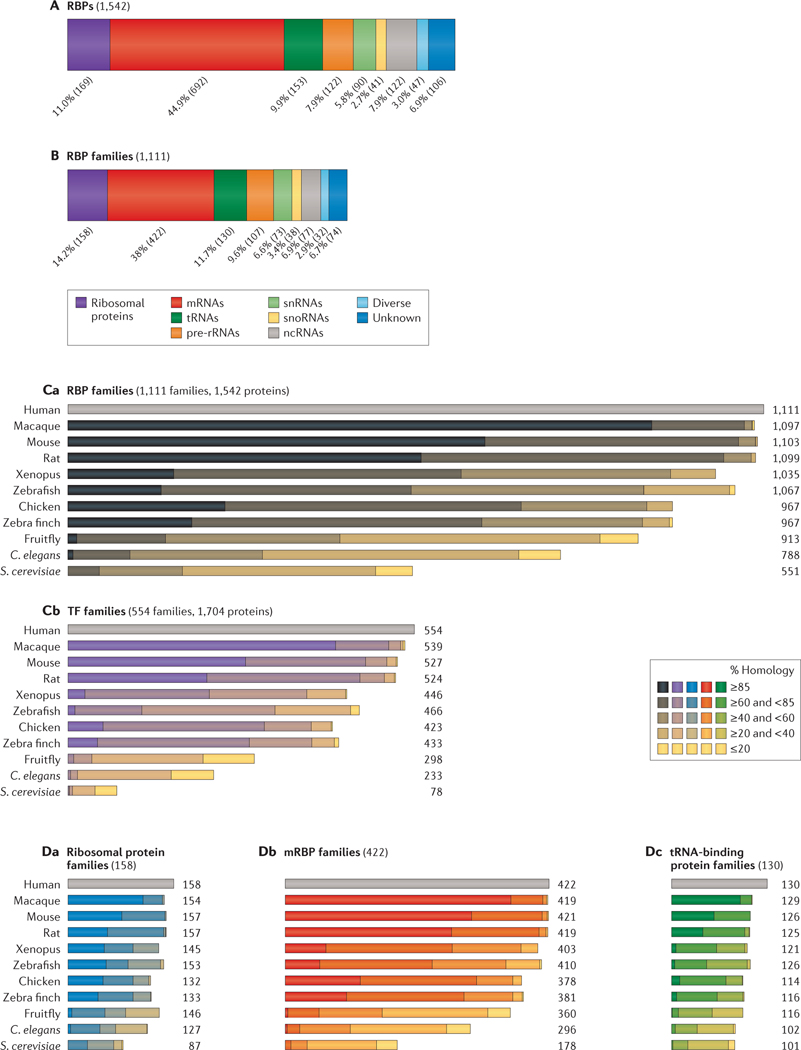
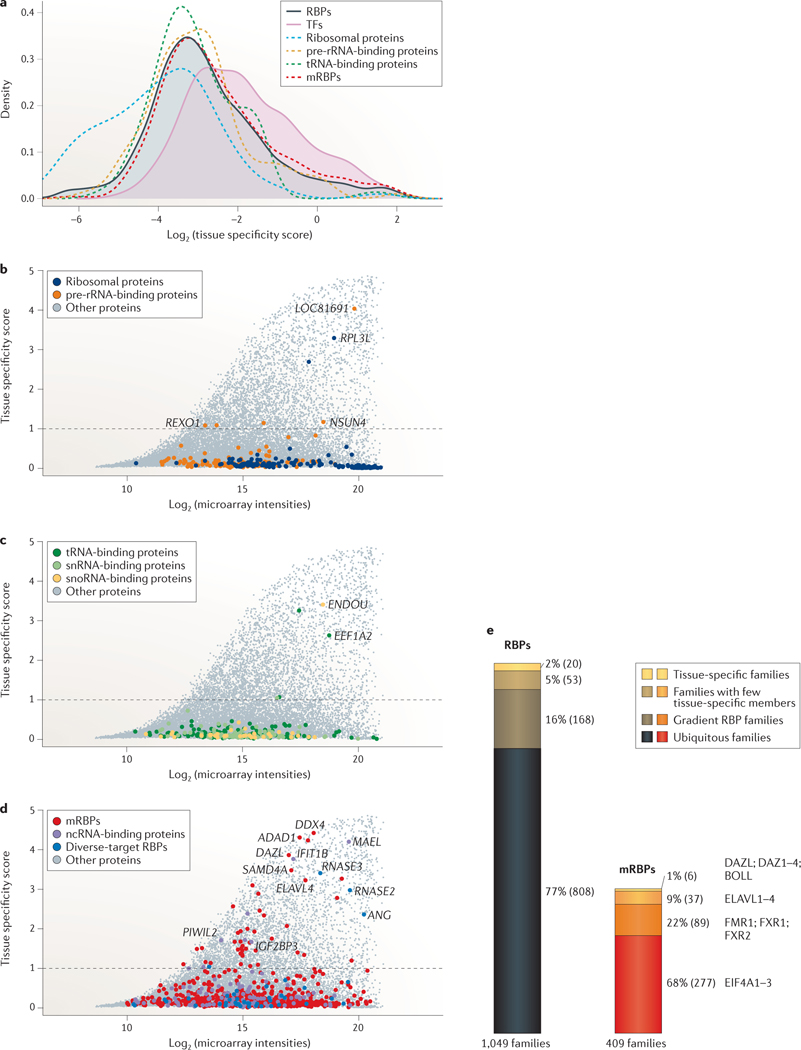
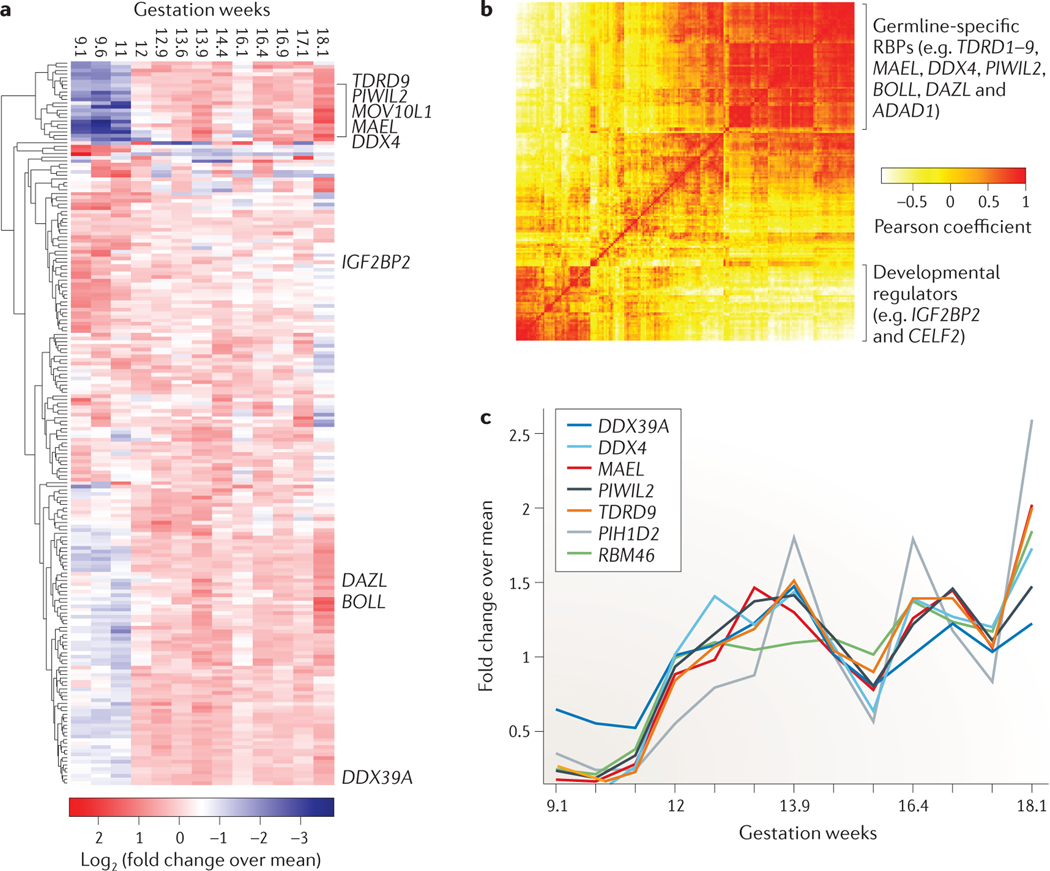
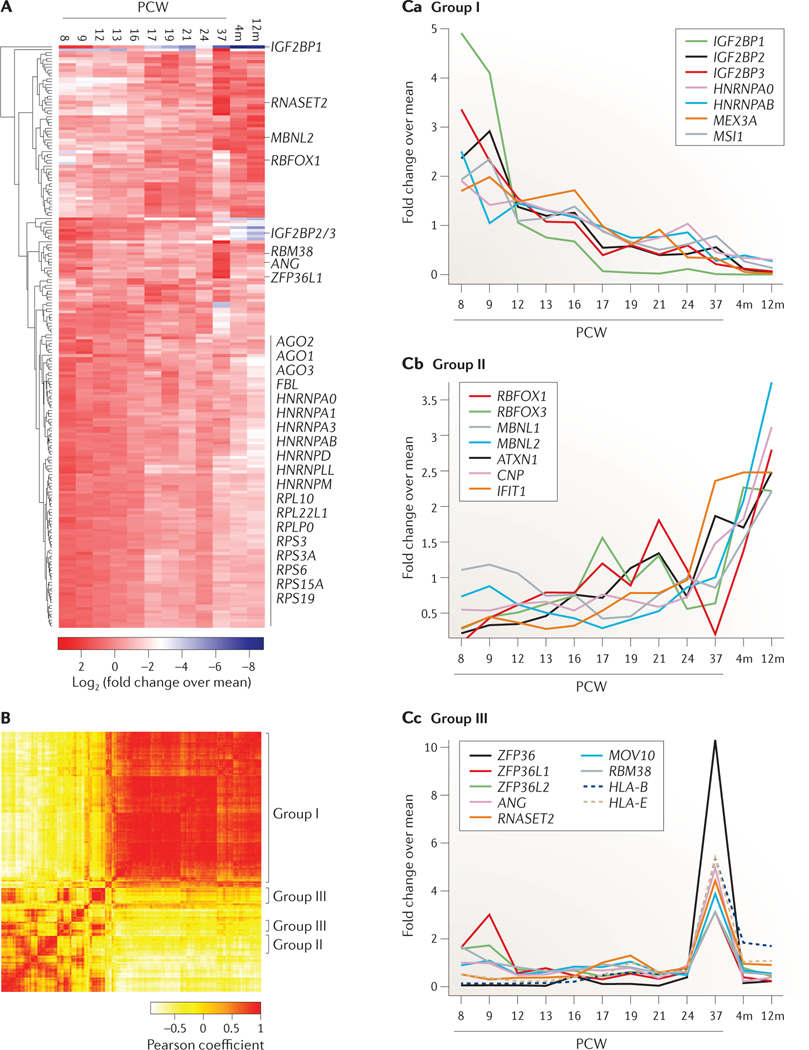
Post-transcriptional gene regulation (PTGR) concerns processes involved in the maturation, transport, stability and translation of coding and non-coding RNAs. RNA-binding proteins (RBPs) and ribonucleoproteins coordinate RNA processing and PTGR. The introduction of large-scale quantitative methods, such as next-generation sequencing and modern protein mass spectrometry, has renewed interest in the investigation of PTGR and the protein factors involved at a systems-biology level. Here, we present a census of 1,542 manually curated RBPs that we have analysed for their interactions with different classes of RNA, their evolutionary conservation, their abundance and their tissue-specific expression. Our analysis is a critical step towards the comprehensive characterization of proteins involved in human RNA metabolism.
Figures
References
-
-
Cech TR & Steitz JA The noncoding RNA revolution — trashing old rules to forge new ones. Cell 157, 77–94 (2014).
This is a concise overview of the different RNA classes in bacteria, archaea and eukaryotes, highlighting their discovery and regulatory roles.
-
-
- Konig J, Zarnack K, Luscombe NM & Ule J. Protein–RNA interactions: new genomic technologies and perspectives. Nature Rev. Genet. 13, 77–83 (2011). - PubMed
-
- Mann M. Functional and quantitative proteomics using SILAC. Nature Rev. Mol. Cell. Biol. 7, 952–958 (2006). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
