

Cellular plasticity induced by anti-α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor encephalitis antibodies
- PMID: 25369168
- PMCID: PMC4365686
- DOI: 10.1002/ana.24293
Cellular plasticity induced by anti-α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor encephalitis antibodies
Abstract
Objective: Autoimmune-mediated anti-α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) encephalitis is a severe but treatment-responsive disorder with prominent short-term memory loss and seizures. The mechanisms by which patient antibodies affect synapses and neurons leading to symptoms are poorly understood.
Methods: The effects of patient antibodies on cultures of live rat hippocampal neurons were determined with immunostaining, Western blot, and electrophysiological analyses.
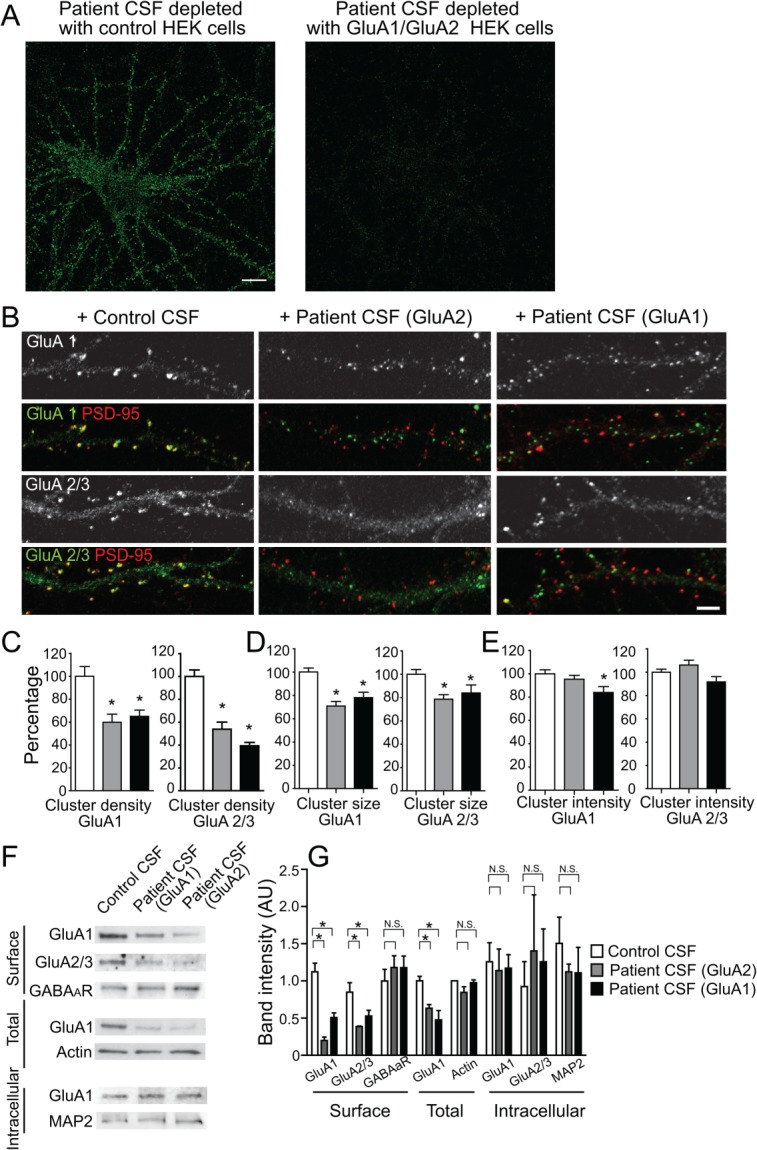
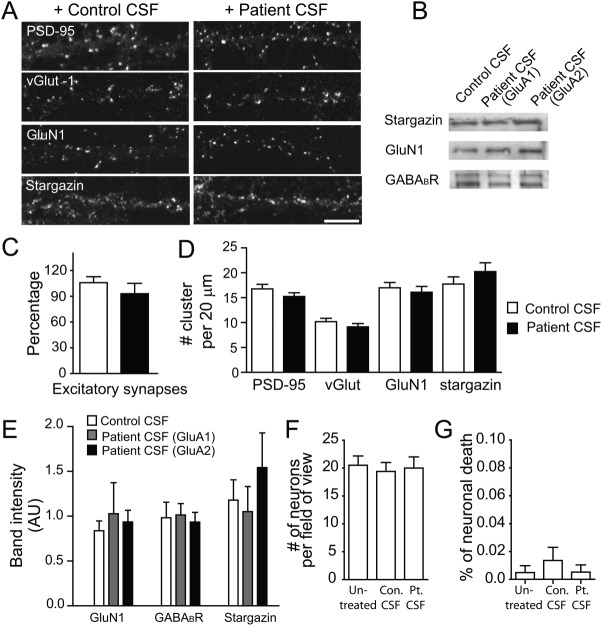
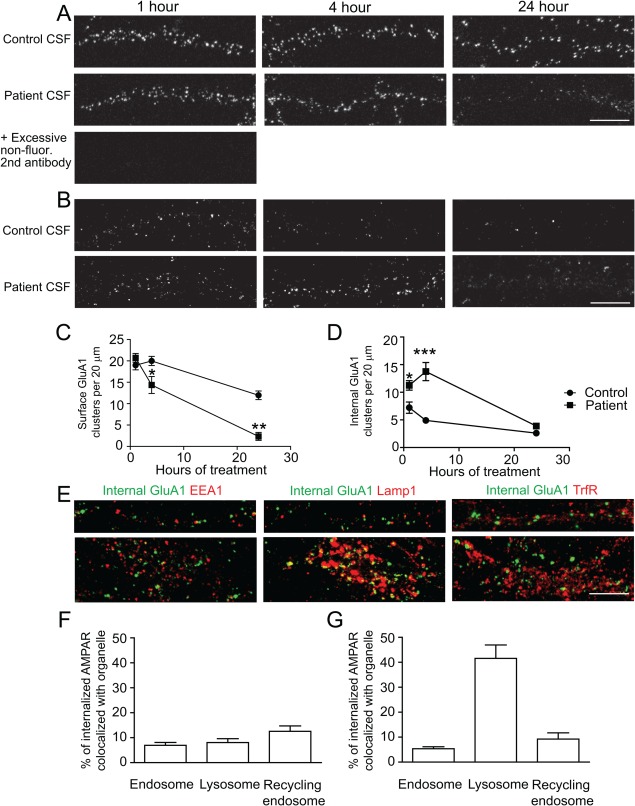
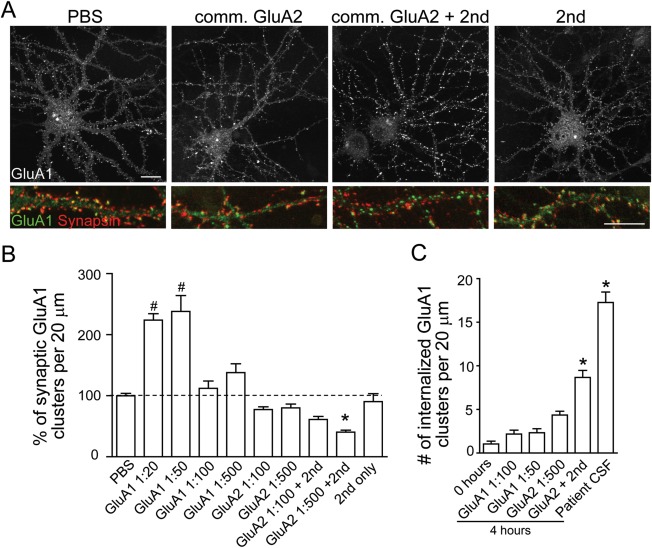
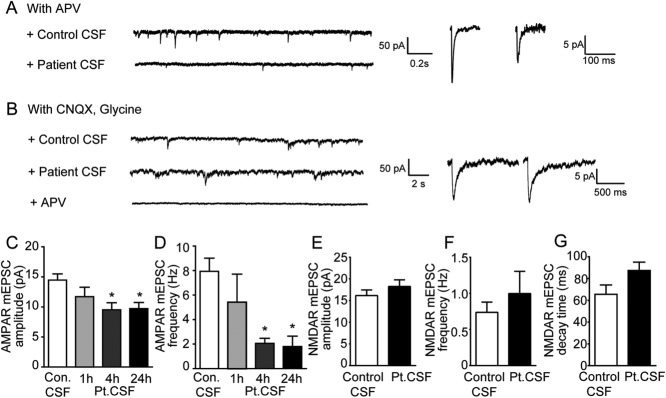
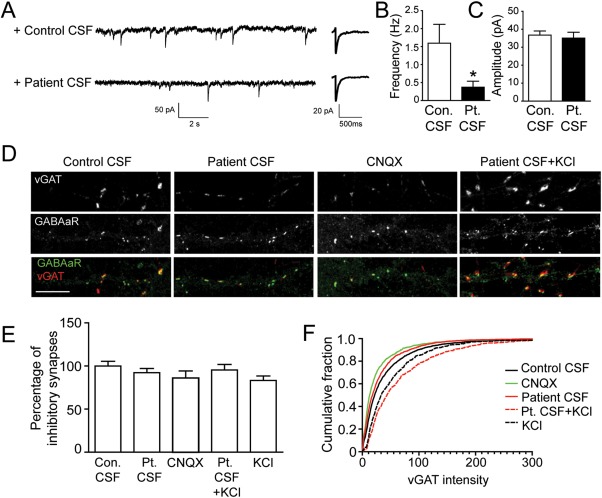
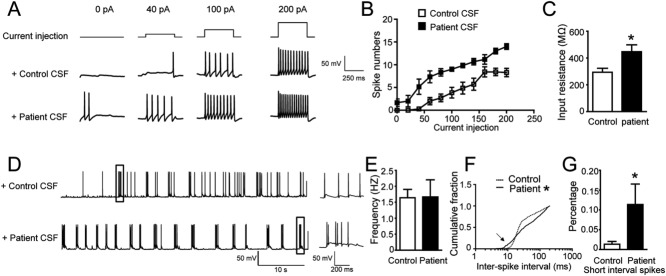
Results: We show that patient antibodies cause a selective decrease in the total surface amount and synaptic localization of GluA1- and GluA2-containing AMPARs, regardless of receptor subunit binding specificity, through increased internalization and degradation of surface AMPAR clusters. In contrast, patient antibodies do not alter the density of excitatory synapses, N-methyl-D-aspartate receptor (NMDAR) clusters, or cell viability. Commercially available AMPAR antibodies directed against extracellular epitopes do not result in a loss of surface and synaptic receptor clusters, suggesting specific effects of patient antibodies. Whole-cell patch clamp recordings of spontaneous miniature postsynaptic currents show that patient antibodies decrease AMPAR-mediated currents, but not NMDAR-mediated currents. Interestingly, several functional properties of neurons are also altered: inhibitory synaptic currents and vesicular γ-aminobutyric acid transporter (vGAT) staining intensity decrease, whereas the intrinsic excitability of neurons and short-interval firing increase.
Interpretation: These results establish that antibodies from patients with anti-AMPAR encephalitis selectively eliminate surface and synaptic AMPARs, resulting in a homeostatic decrease in inhibitory synaptic transmission and increased intrinsic excitability, which may contribute to the memory deficits and epilepsy that are prominent in patients with this disorder.
© 2014 The Authors Annals of Neurology published by Wiley Periodicals, Inc. on behalf of American Neurological Association.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
