

Genomic epidemiology of the Haitian cholera outbreak: a single introduction followed by rapid, extensive, and continued spread characterized the onset of the epidemic
- PMID: 25370488
- PMCID: PMC4222100
- DOI: 10.1128/mBio.01721-14
Genomic epidemiology of the Haitian cholera outbreak: a single introduction followed by rapid, extensive, and continued spread characterized the onset of the epidemic
Abstract
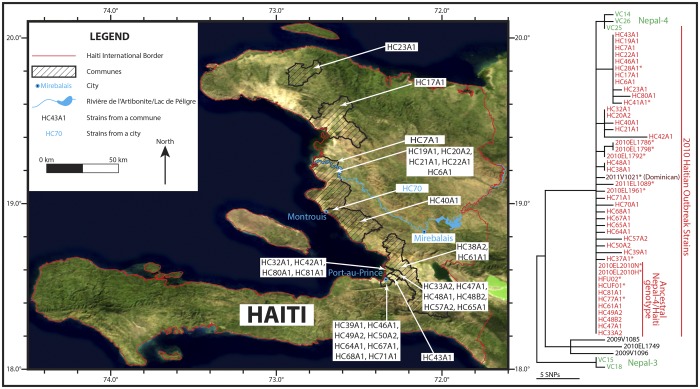
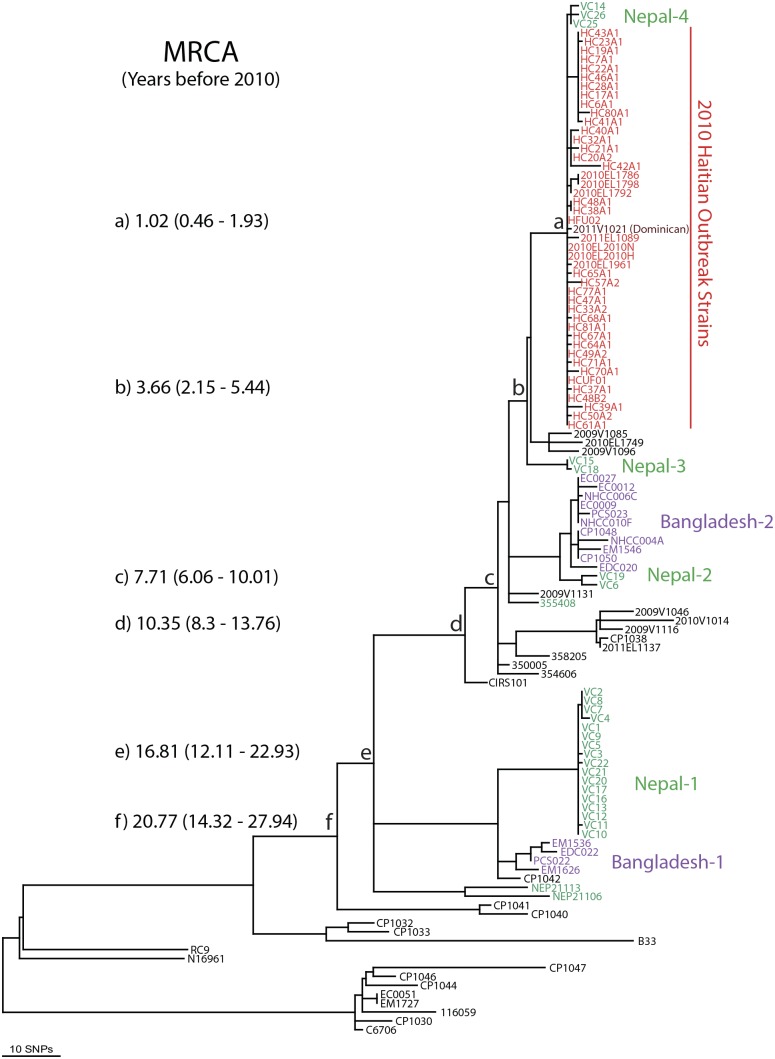
For centuries, cholera has been one of the most feared diseases. The causative agent Vibrio cholerae is a waterborne Gram-negative enteric pathogen eliciting a severe watery diarrheal disease. In October 2010, the seventh pandemic reached Haiti, a country that had not experienced cholera for more than a century. By using whole-genome sequence typing and mapping strategies of 116 serotype O1 strains from global sources, including 44 Haitian genomes, we present a detailed reconstructed evolutionary history of the seventh pandemic with a focus on the Haitian outbreak. We catalogued subtle genomic alterations at the nucleotide level in the genome core and architectural rearrangements from whole-genome map comparisons. Isolates closely related to the Haitian isolates caused several recent outbreaks in southern Asia. This study provides evidence for a single-source introduction of cholera from Nepal into Haiti followed by rapid, extensive, and continued clonal expansion. The phylogeographic patterns in both southern Asia and Haiti argue for the rapid dissemination of V. cholerae across the landscape necessitating real-time surveillance efforts to complement the whole-genome epidemiological analysis. As eradication efforts move forward, phylogeographic knowledge will be important for identifying persistent sources and monitoring success at regional levels. The results of molecular and epidemiological analyses of this outbreak suggest that an indigenous Haitian source of V. cholerae is unlikely and that an indigenous source has not contributed to the genomic evolution of this clade.
Importance: In this genomic epidemiology study, we have applied high-resolution whole-genome-based sequence typing methodologies on a comprehensive set of genome sequences that have become available in the aftermath of the Haitian cholera epidemic. These sequence resources enabled us to reassess the degree of genomic heterogeneity within the Vibrio cholerae O1 serotype and to refine boundaries and evolutionary relationships. The established phylogenomic framework showed how outbreak isolates fit into the global phylogeographic patterns compared to a comprehensive globally and temporally diverse strain collection and provides strong molecular evidence that points to a nonindigenous source of the 2010 Haitian cholera outbreak and refines epidemiological standards used in outbreak investigations for outbreak inclusion/exclusion following the concept of genomic epidemiology. The generated phylogenomic data have major public health relevance in translating sequence-based information to assist in future diagnostic, epidemiological, surveillance, and forensic studies of cholera.
Copyright © 2014 Eppinger et al.
Figures
Similar articles
-
Deciphering the origins and tracking the evolution of cholera epidemics with whole-genome-based molecular epidemiology.mBio. 2013 Sep 10;4(5):e00670-13. doi: 10.1128/mBio.00670-13. mBio. 2013. PMID: 24023387 Free PMC article.
-
Evolutionary dynamics of Vibrio cholerae O1 following a single-source introduction to Haiti.mBio. 2013 Jul 2;4(4):e00398-13. doi: 10.1128/mBio.00398-13. mBio. 2013. PMID: 23820394 Free PMC article.
-
Hybrid Vibrio cholerae El Tor lacking SXT identified as the cause of a cholera outbreak in the Philippines.mBio. 2015 Apr 21;6(2):e00047-15. doi: 10.1128/mBio.00047-15. mBio. 2015. PMID: 25900650 Free PMC article.
-
Widespread epidemic cholera caused by a restricted subset of Vibrio cholerae clones.Clin Microbiol Infect. 2014 May;20(5):373-9. doi: 10.1111/1469-0691.12610. Epub 2014 Mar 29. Clin Microbiol Infect. 2014. PMID: 24575898 Review.
-
Whole-genome sequence comparisons reveal the evolution of Vibrio cholerae O1.Trends Microbiol. 2015 Aug;23(8):479-89. doi: 10.1016/j.tim.2015.03.010. Epub 2015 Apr 24. Trends Microbiol. 2015. PMID: 25913612 Review.
Cited by
-
On the predictive ability of mechanistic models for the Haitian cholera epidemic.J R Soc Interface. 2015 Mar 6;12(104):20140840. doi: 10.1098/rsif.2014.0840. J R Soc Interface. 2015. PMID: 25631563 Free PMC article.
-
WGS Data Collections: How Do Genomic Databases Transform Medicine?Int J Mol Sci. 2023 Feb 3;24(3):3031. doi: 10.3390/ijms24033031. Int J Mol Sci. 2023. PMID: 36769353 Free PMC article. Review.
-
Genomes of Vibrio metoecus co-isolated with Vibrio cholerae extend our understanding of differences between these closely related species.Gut Pathog. 2022 Nov 20;14(1):42. doi: 10.1186/s13099-022-00516-x. Gut Pathog. 2022. PMID: 36404338 Free PMC article.
-
Microbe hunting in the modern era: reflecting on a decade of microbial genomic epidemiology.Curr Biol. 2020 Oct 5;30(19):R1124-R1130. doi: 10.1016/j.cub.2020.06.097. Curr Biol. 2020. PMID: 33022254 Free PMC article.
-
Salmonella enterica Serovar Typhi in Bangladesh: Exploration of Genomic Diversity and Antimicrobial Resistance.mBio. 2018 Nov 13;9(6):e02112-18. doi: 10.1128/mBio.02112-18. mBio. 2018. PMID: 30425150 Free PMC article.
References
-
- Mutreja A, Kim DW, Thomson NR, Connor TR, Lee JH, Kariuki S, Croucher NJ, Choi SY, Harris SR, Lebens M, Niyogi SK, Kim EJ, Ramamurthy T, Chun J, Wood JL, Clemens JD, Czerkinsky C, Nair GB, Holmgren J, Parkhill J, Dougan G. 2011. Evidence for several waves of global transmission in the seventh cholera pandemic. Nature 477:462–465. 10.1038/nature10392. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
