

Increased BDNF protein expression after ischemic or PKC epsilon preconditioning promotes electrophysiologic changes that lead to neuroprotection
- PMID: 25370861
- PMCID: PMC4294405
- DOI: 10.1038/jcbfm.2014.185
Increased BDNF protein expression after ischemic or PKC epsilon preconditioning promotes electrophysiologic changes that lead to neuroprotection
Abstract
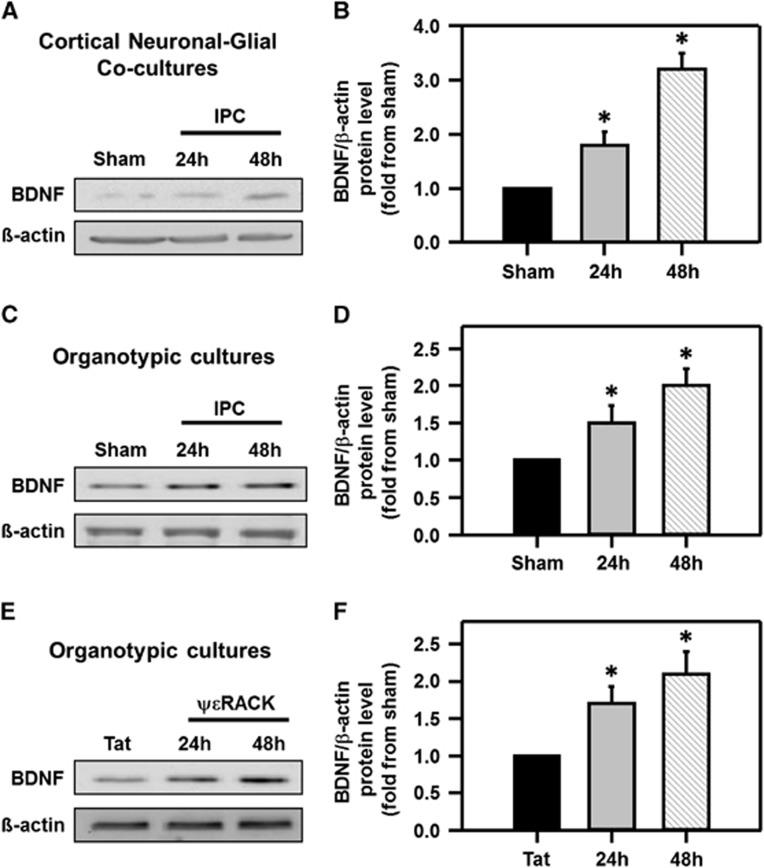
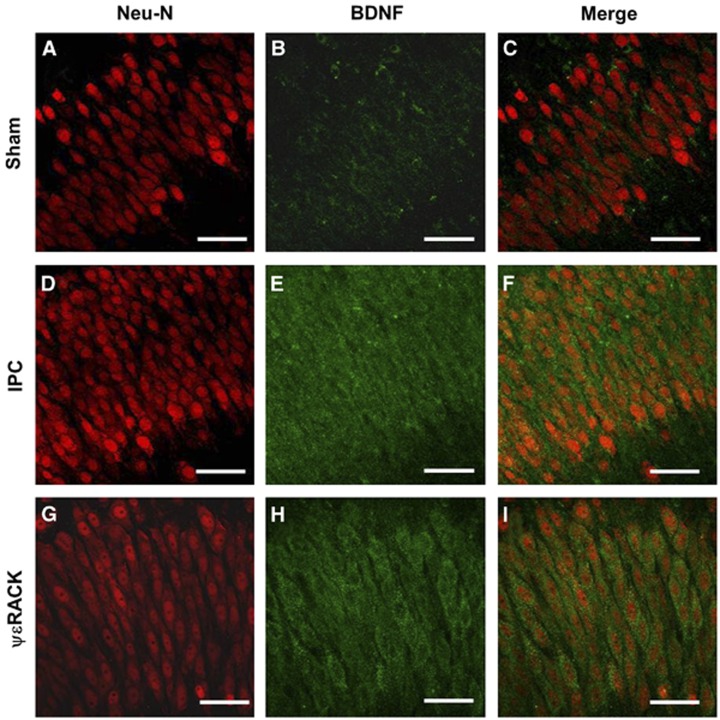
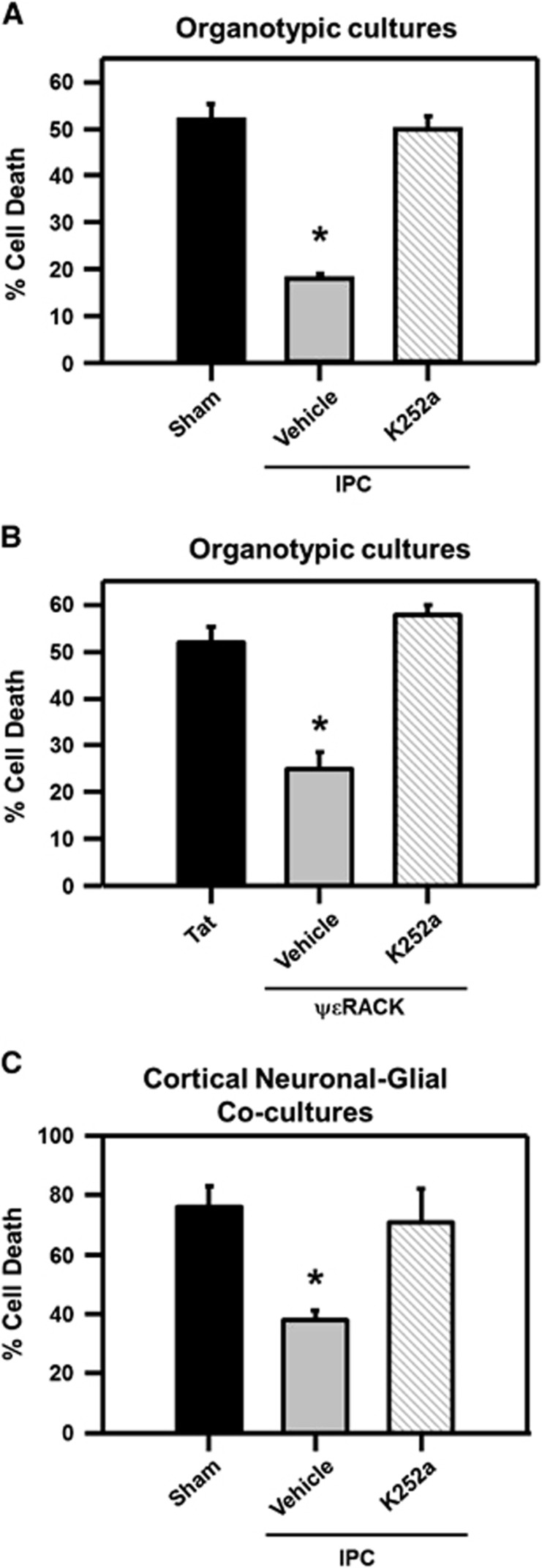
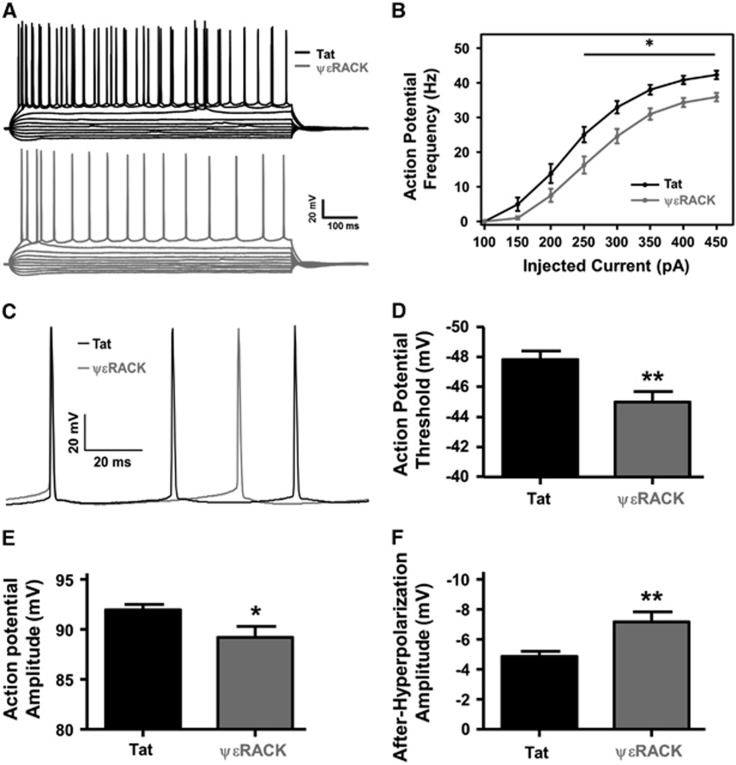
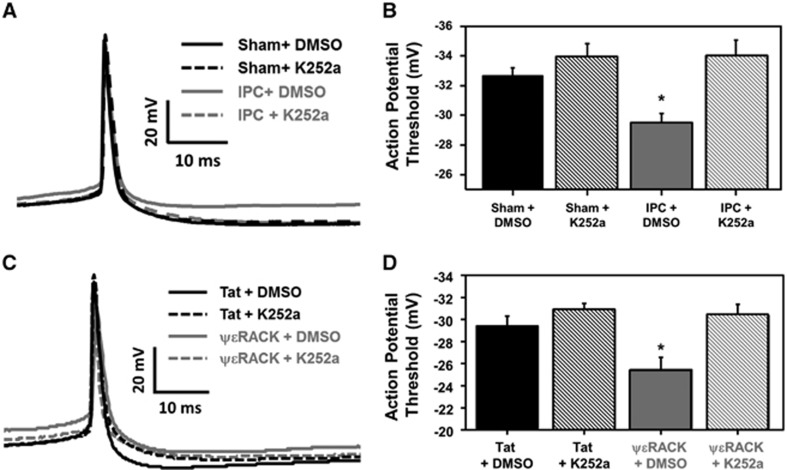
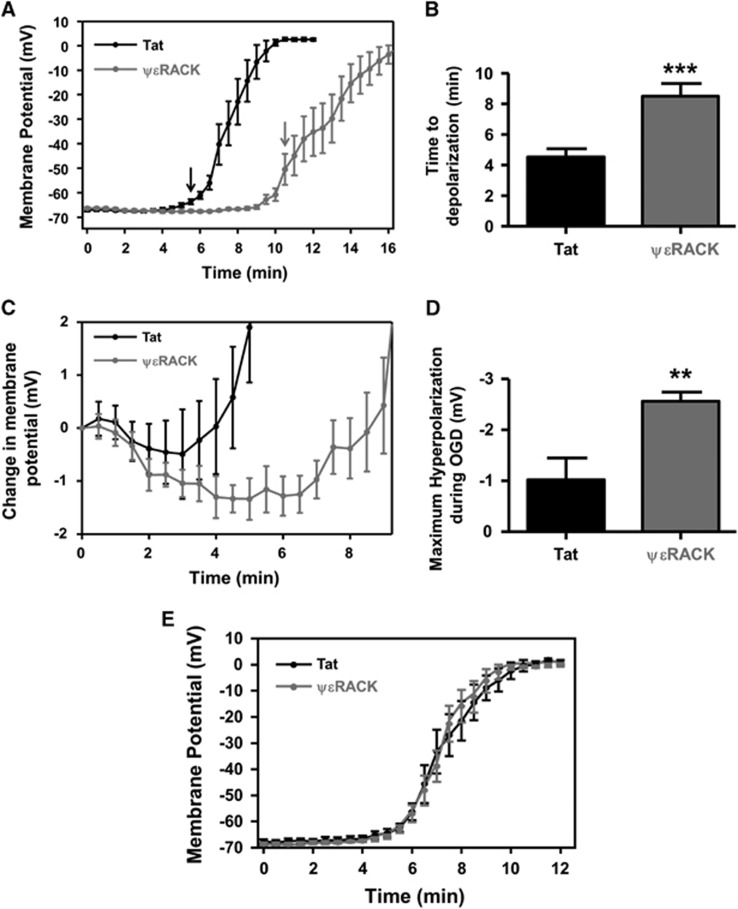
Ischemic preconditioning (IPC) via protein kinase C epsilon (PKCɛ) activation induces neuroprotection against lethal ischemia. Brain-derived neurotrophic factor (BDNF) is a pro-survival signaling molecule that modulates synaptic plasticity and neurogenesis. Interestingly, BDNF mRNA expression increases after IPC. In this study, we investigated whether IPC or pharmacological preconditioning (PKCɛ activation) promoted BDNF-induced neuroprotection, if neuroprotection by IPC or PKCɛ activation altered neuronal excitability, and whether these changes were BDNF-mediated. We used both in vitro (hippocampal organotypic cultures and cortical neuronal-glial cocultures) and in vivo (acute hippocampal slices 48 hours after preconditioning) models of IPC or PKCɛ activation. BDNF protein expression increased 24 to 48 hours after preconditioning, where inhibition of the BDNF Trk receptors abolished neuroprotection against oxygen and glucose deprivation (OGD) in vitro. In addition, there was a significant decrease in neuronal firing frequency and increase in threshold potential 48 hours after preconditioning in vivo, where this threshold modulation was dependent on BDNF activation of Trk receptors in excitatory cortical neurons. In addition, 48 hours after PKCɛ activation in vivo, the onset of anoxic depolarization during OGD was significantly delayed in hippocampal slices. Overall, these results suggest that after IPC or PKCɛ activation, there are BDNF-dependent electrophysiologic modifications that lead to neuroprotection.
Figures
References
-
- Nichols-Larsen DS, Clark PC, Zeringue A, Greenspan A, Blanton S. Factors influencing stroke survivors' quality of life during subacute recovery. Stroke. 2005;36:1480–1484. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
