

Large transcription units unify copy number variants and common fragile sites arising under replication stress
- PMID: 25373142
- PMCID: PMC4315293
- DOI: 10.1101/gr.177121.114
Large transcription units unify copy number variants and common fragile sites arising under replication stress
Abstract
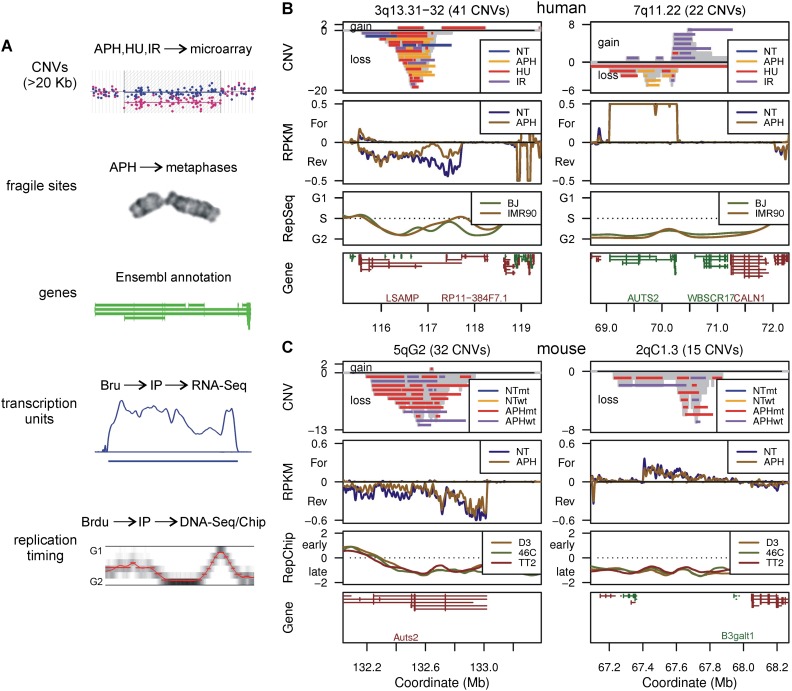
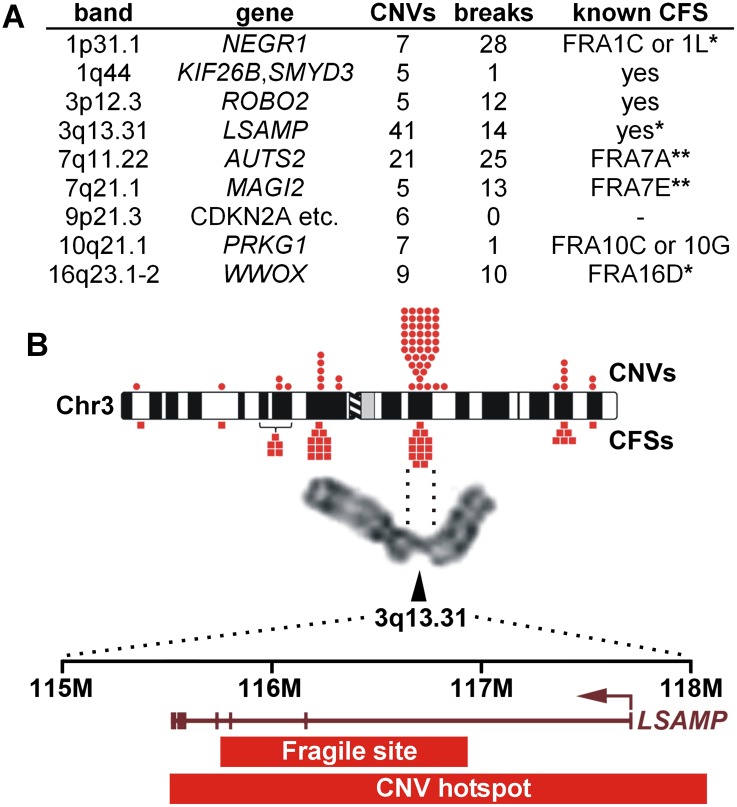
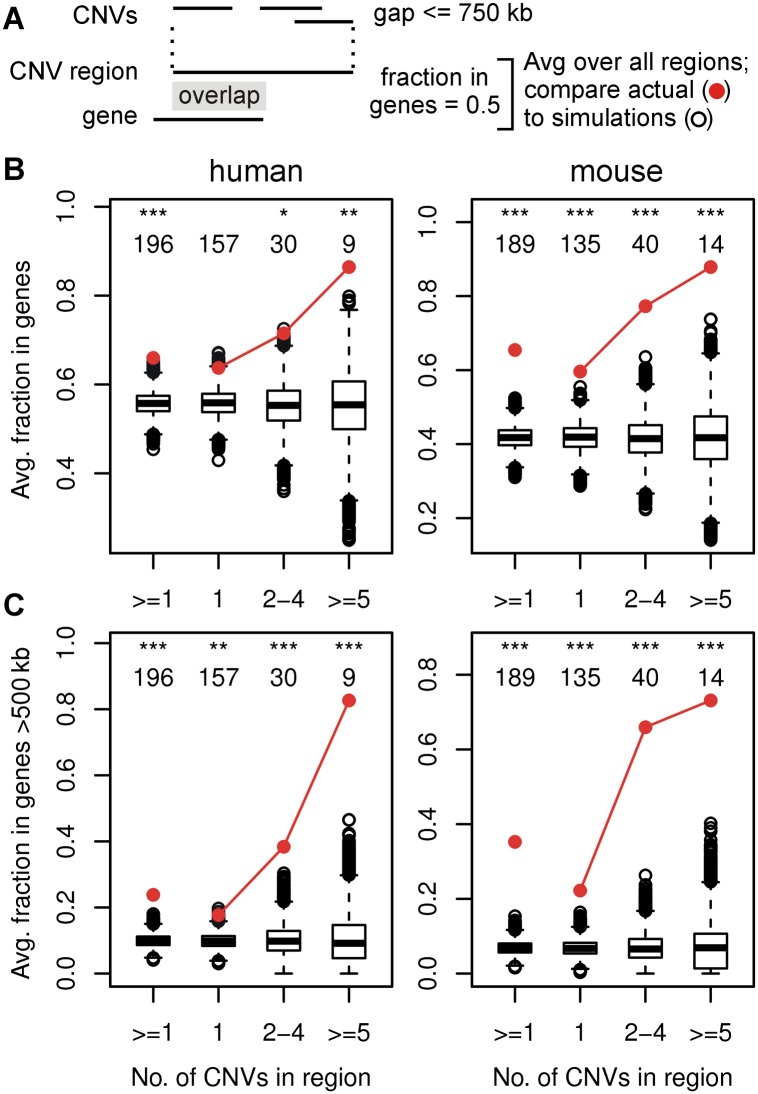
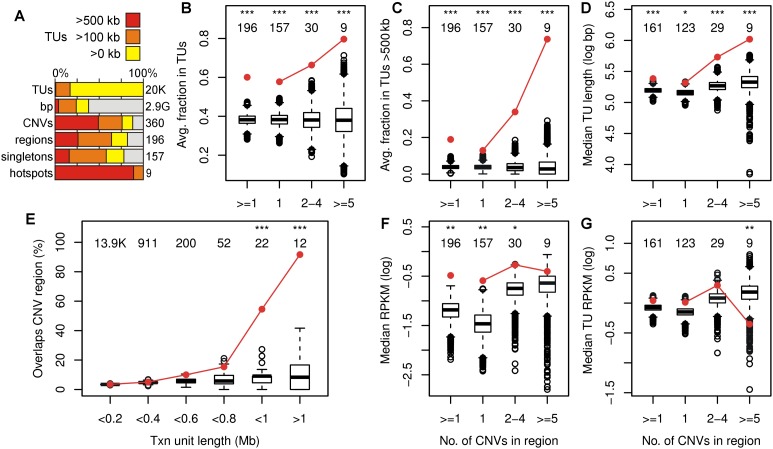
Copy number variants (CNVs) resulting from genomic deletions and duplications and common fragile sites (CFSs) seen as breaks on metaphase chromosomes are distinct forms of structural chromosome instability precipitated by replication inhibition. Although they share a common induction mechanism, it is not known how CNVs and CFSs are related or why some genomic loci are much more prone to their occurrence. Here we compare large sets of de novo CNVs and CFSs in several experimental cell systems to each other and to overlapping genomic features. We first show that CNV hotpots and CFSs occurred at the same human loci within a given cultured cell line. Bru-seq nascent RNA sequencing further demonstrated that although genomic regions with low CNV frequencies were enriched in transcribed genes, the CNV hotpots that matched CFSs specifically corresponded to the largest active transcription units in both human and mouse cells. Consistently, active transcription units >1 Mb were robust cell-type-specific predictors of induced CNV hotspots and CFS loci. Unlike most transcribed genes, these very large transcription units replicated late and organized deletion and duplication CNVs into their transcribed and flanking regions, respectively, supporting a role for transcription in replication-dependent lesion formation. These results indicate that active large transcription units drive extreme locus- and cell-type-specific genomic instability under replication stress, resulting in both CNVs and CFSs as different manifestations of perturbed replication dynamics.
© 2015 Wilson et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
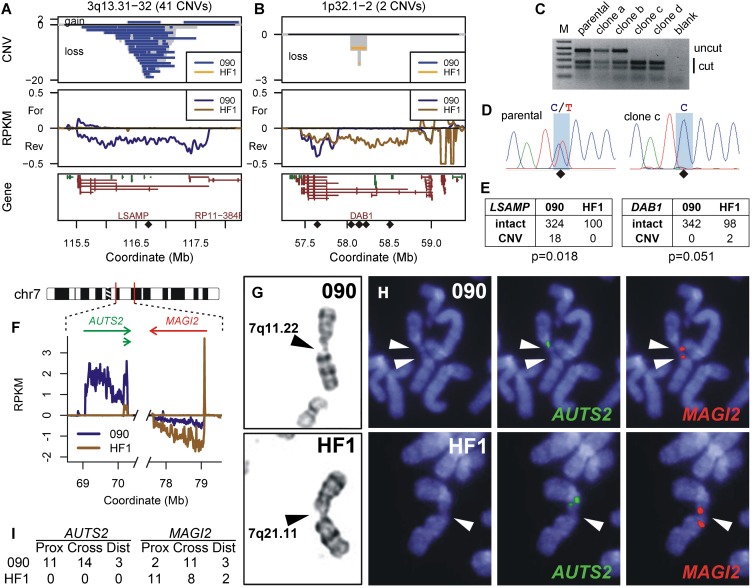
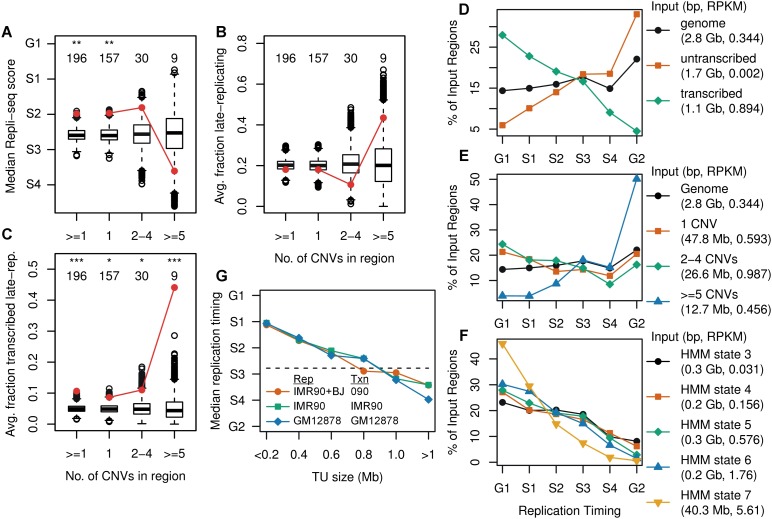
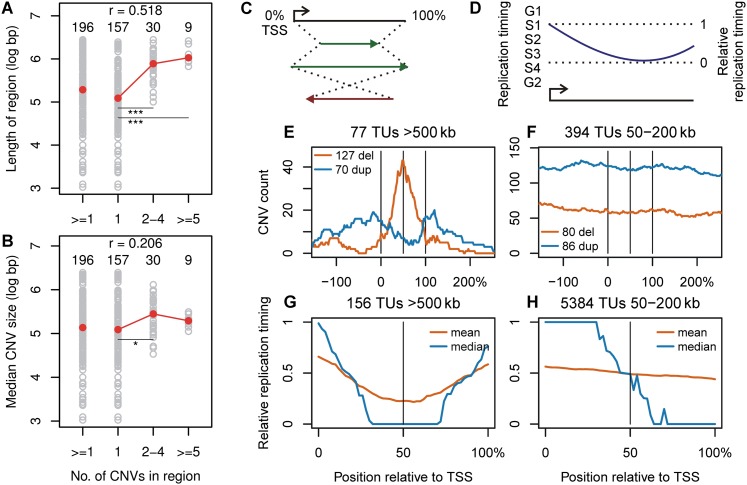
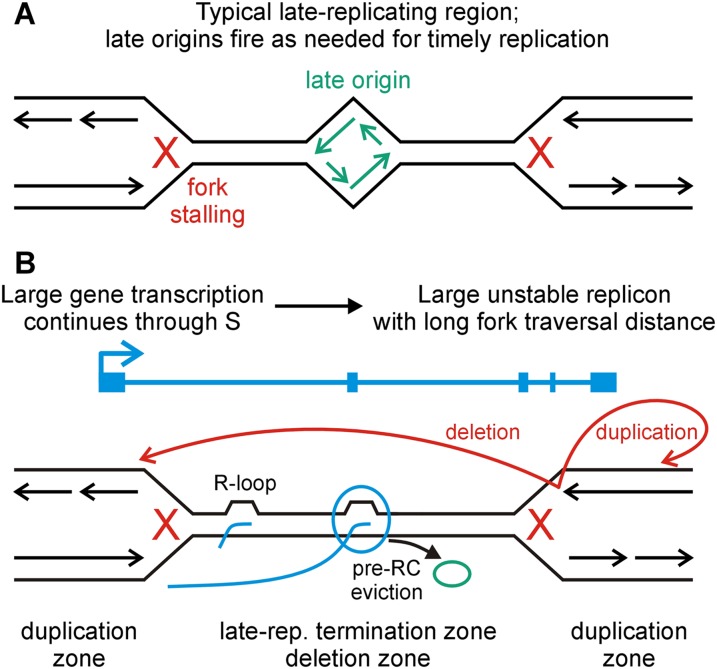
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous