

Single haplotype assembly of the human genome from a hydatidiform mole
- PMID: 25373144
- PMCID: PMC4248323
- DOI: 10.1101/gr.180893.114
Single haplotype assembly of the human genome from a hydatidiform mole
Abstract
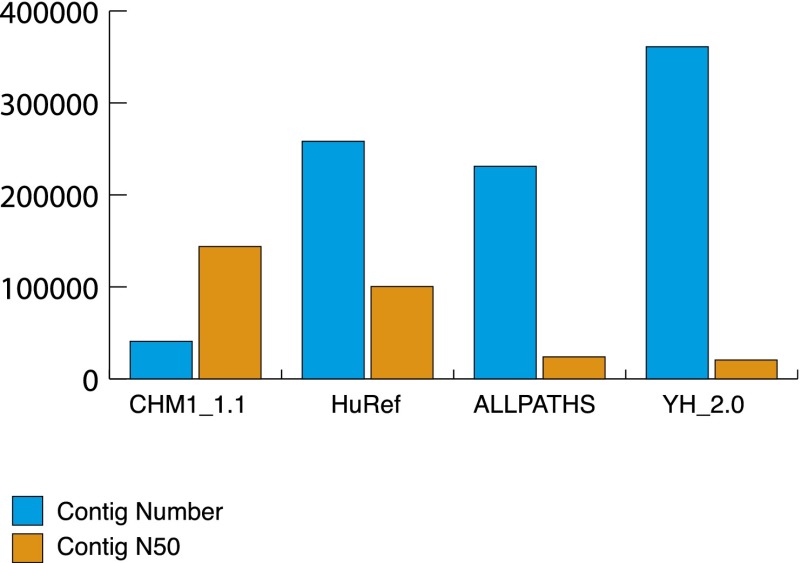
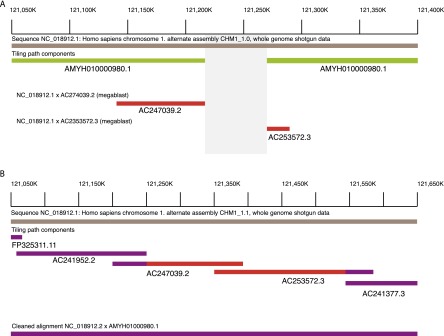
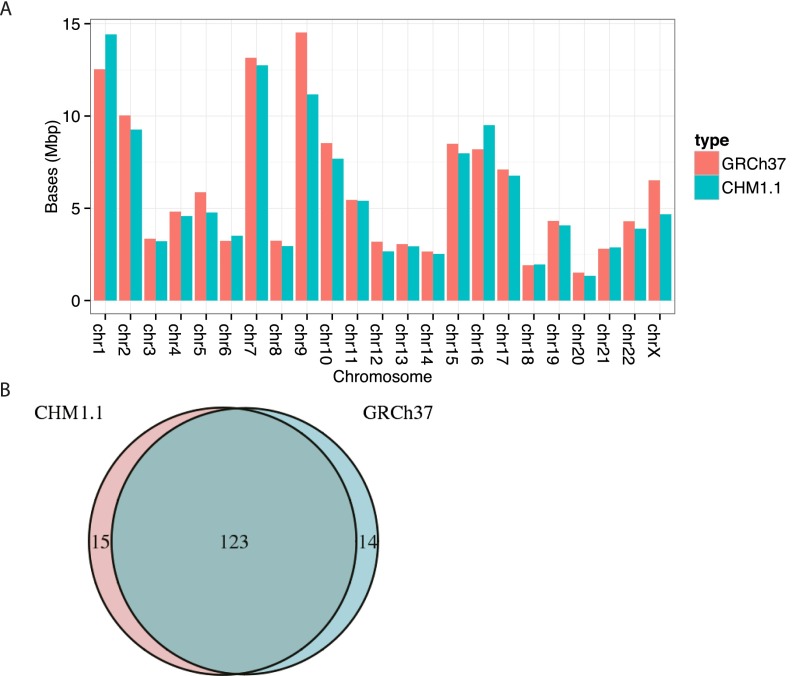
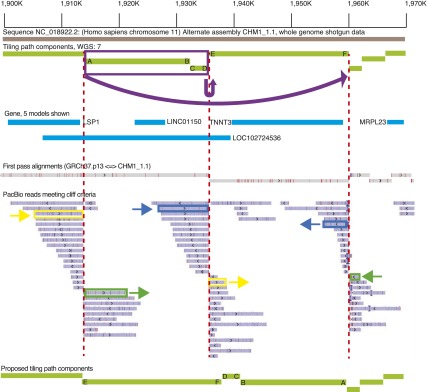
A complete reference assembly is essential for accurately interpreting individual genomes and associating variation with phenotypes. While the current human reference genome sequence is of very high quality, gaps and misassemblies remain due to biological and technical complexities. Large repetitive sequences and complex allelic diversity are the two main drivers of assembly error. Although increasing the length of sequence reads and library fragments can improve assembly, even the longest available reads do not resolve all regions. In order to overcome the issue of allelic diversity, we used genomic DNA from an essentially haploid hydatidiform mole, CHM1. We utilized several resources from this DNA including a set of end-sequenced and indexed BAC clones and 100× Illumina whole-genome shotgun (WGS) sequence coverage. We used the WGS sequence and the GRCh37 reference assembly to create an assembly of the CHM1 genome. We subsequently incorporated 382 finished BAC clone sequences to generate a draft assembly, CHM1_1.1 (NCBI AssemblyDB GCA_000306695.2). Analysis of gene, repetitive element, and segmental duplication content show this assembly to be of excellent quality and contiguity. However, comparison to assembly-independent resources, such as BAC clone end sequences and PacBio long reads, indicate misassembled regions. Most of these regions are enriched for structural variation and segmental duplication, and can be resolved in the future. This publicly available assembly will be integrated into the Genome Reference Consortium curation framework for further improvement, with the ultimate goal being a completely finished gap-free assembly.
© 2014 Steinberg et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
-
- Barbouti A, Stankiewicz P, Nusbaum C, Cuomo C, Cook A, Hoglund M, Johansson B, Hagemeijer A, Park SS, Mitelman F, et al. . 2004. The breakpoint region of the most common isochromosome, i(17q), in human neoplasia is characterized by a complex genomic architecture with large, palindromic, low-copy repeats. Am J Hum Genet 74: 1–10. - PMC - PubMed
Publication types
MeSH terms
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous