

Genome-wide associations between genetic and epigenetic variation influence mRNA expression and insulin secretion in human pancreatic islets
- PMID: 25375650
- PMCID: PMC4222689
- DOI: 10.1371/journal.pgen.1004735
Genome-wide associations between genetic and epigenetic variation influence mRNA expression and insulin secretion in human pancreatic islets
Erratum in
- PLoS Genet. 2014 Dec;10(12):e1004886
Abstract
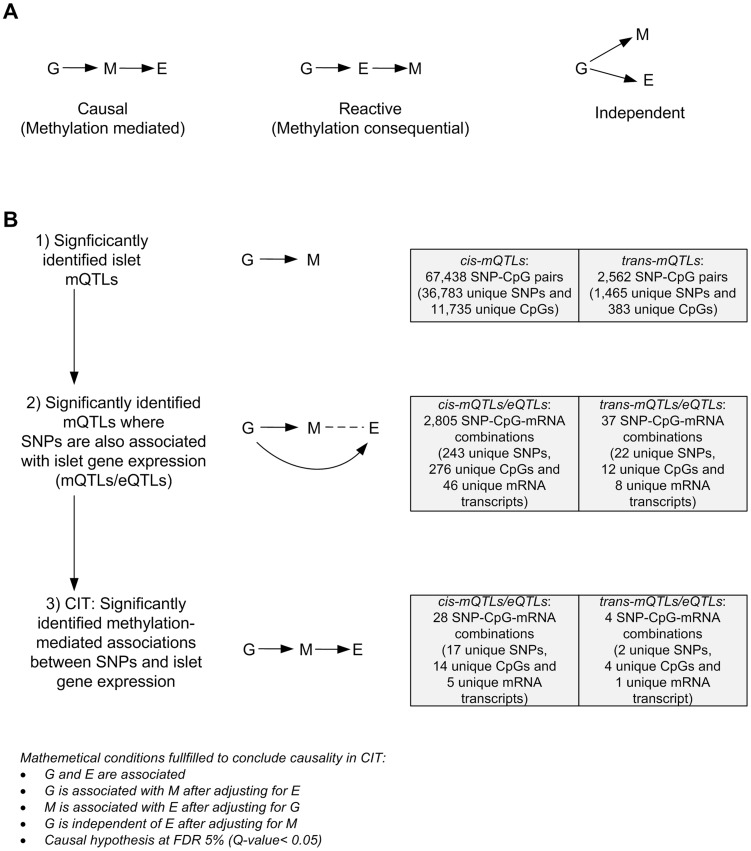
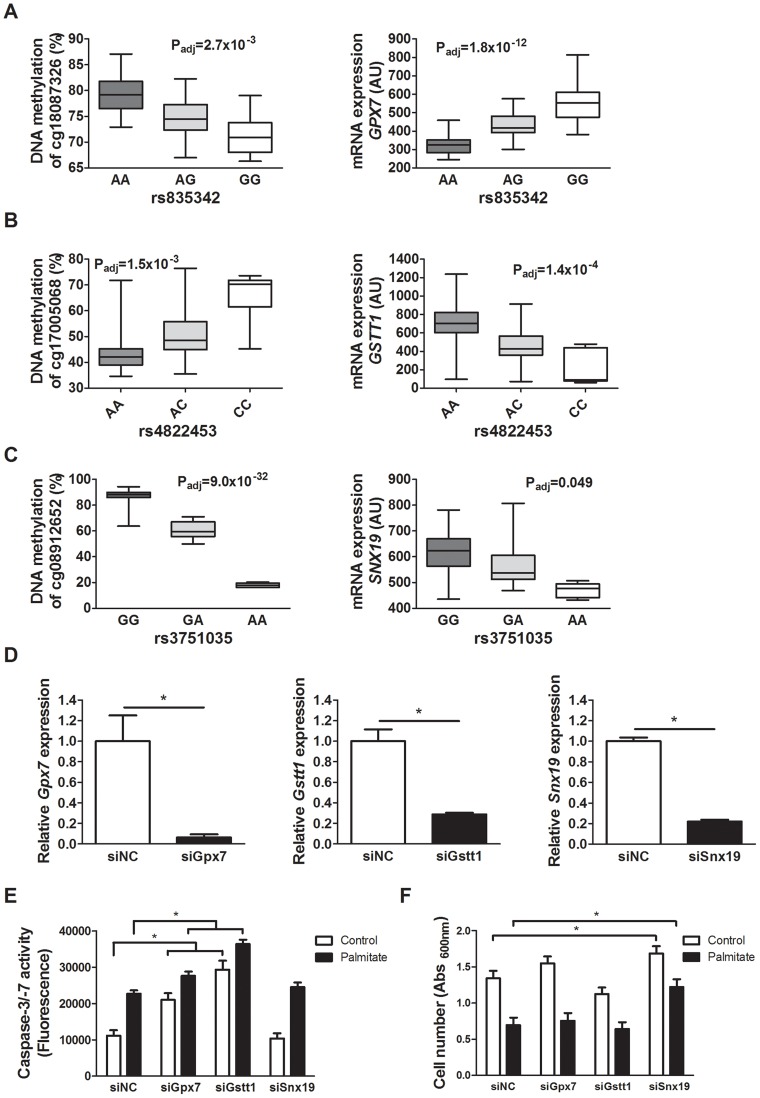
Genetic and epigenetic mechanisms may interact and together affect biological processes and disease development. However, most previous studies have investigated genetic and epigenetic mechanisms independently, and studies examining their interactions throughout the human genome are lacking. To identify genetic loci that interact with the epigenome, we performed the first genome-wide DNA methylation quantitative trait locus (mQTL) analysis in human pancreatic islets. We related 574,553 single nucleotide polymorphisms (SNPs) with genome-wide DNA methylation data of 468,787 CpG sites targeting 99% of RefSeq genes in islets from 89 donors. We identified 67,438 SNP-CpG pairs in cis, corresponding to 36,783 SNPs (6.4% of tested SNPs) and 11,735 CpG sites (2.5% of tested CpGs), and 2,562 significant SNP-CpG pairs in trans, corresponding to 1,465 SNPs (0.3% of tested SNPs) and 383 CpG sites (0.08% of tested CpGs), showing significant associations after correction for multiple testing. These include reported diabetes loci, e.g. ADCY5, KCNJ11, HLA-DQA1, INS, PDX1 and GRB10. CpGs of significant cis-mQTLs were overrepresented in the gene body and outside of CpG islands. Follow-up analyses further identified mQTLs associated with gene expression and insulin secretion in human islets. Causal inference test (CIT) identified SNP-CpG pairs where DNA methylation in human islets is the potential mediator of the genetic association with gene expression or insulin secretion. Functional analyses further demonstrated that identified candidate genes (GPX7, GSTT1 and SNX19) directly affect key biological processes such as proliferation and apoptosis in pancreatic β-cells. Finally, we found direct correlations between DNA methylation of 22,773 (4.9%) CpGs with mRNA expression of 4,876 genes, where 90% of the correlations were negative when CpGs were located in the region surrounding transcription start site. Our study demonstrates for the first time how genome-wide genetic and epigenetic variation interacts to influence gene expression, islet function and potential diabetes risk in humans.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures






Similar articles
-
A Genome-Wide mQTL Analysis in Human Adipose Tissue Identifies Genetic Variants Associated with DNA Methylation, Gene Expression and Metabolic Traits.PLoS One. 2016 Jun 20;11(6):e0157776. doi: 10.1371/journal.pone.0157776. eCollection 2016. PLoS One. 2016. PMID: 27322064 Free PMC article.
-
Genome-wide DNA methylation analysis of human pancreatic islets from type 2 diabetic and non-diabetic donors identifies candidate genes that influence insulin secretion.PLoS Genet. 2014 Mar 6;10(3):e1004160. doi: 10.1371/journal.pgen.1004160. eCollection 2014 Mar. PLoS Genet. 2014. PMID: 24603685 Free PMC article.
-
Identification of CpG-SNPs associated with type 2 diabetes and differential DNA methylation in human pancreatic islets.Diabetologia. 2013 May;56(5):1036-46. doi: 10.1007/s00125-012-2815-7. Epub 2013 Mar 6. Diabetologia. 2013. PMID: 23462794 Free PMC article.
-
Epigenetic regulation of insulin action and secretion - role in the pathogenesis of type 2 diabetes.J Intern Med. 2020 Aug;288(2):158-167. doi: 10.1111/joim.13049. Epub 2020 May 3. J Intern Med. 2020. PMID: 32363639 Review.
-
Does epigenetic dysregulation of pancreatic islets contribute to impaired insulin secretion and type 2 diabetes?Biochem Cell Biol. 2015 Oct;93(5):511-21. doi: 10.1139/bcb-2015-0057. Epub 2015 Aug 4. Biochem Cell Biol. 2015. PMID: 26369706 Review.
Cited by
-
Nutritional systems biology of type 2 diabetes.Genes Nutr. 2015 Sep;10(5):481. doi: 10.1007/s12263-015-0481-3. Epub 2015 Jul 24. Genes Nutr. 2015. PMID: 26202330 Free PMC article.
-
DNA methylation: a cause and consequence of type 2 diabetes.Genomics Inform. 2019 Dec;17(4):e38. doi: 10.5808/GI.2019.17.4.e38. Epub 2019 Nov 28. Genomics Inform. 2019. PMID: 31896238 Free PMC article. Review.
-
Interaction of the sorting nexin 25 homologue Snazarus with Rab11 balances endocytic and secretory transport and maintains the ultrafiltration diaphragm in nephrocytes.Mol Biol Cell. 2023 Aug 1;34(9):ar87. doi: 10.1091/mbc.E22-09-0421. Epub 2023 Jun 14. Mol Biol Cell. 2023. PMID: 37314856 Free PMC article.
-
Analyzing Gene Expression After Administration of Low-Intensity Therapeutic Ultrasound in Human Islet Cells.J Ultrasound Med. 2024 Jun;43(6):1131-1141. doi: 10.1002/jum.16441. Epub 2024 Feb 27. J Ultrasound Med. 2024. PMID: 38414281 Free PMC article.
-
DNA methylation of loci within ABCG1 and PHOSPHO1 in blood DNA is associated with future type 2 diabetes risk.Epigenetics. 2016 Jul 2;11(7):482-8. doi: 10.1080/15592294.2016.1178418. Epub 2016 May 5. Epigenetics. 2016. PMID: 27148772 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
