

Optimization of transcription factor binding map accuracy utilizing knockout-mouse models
- PMID: 25378309
- PMCID: PMC4245947
- DOI: 10.1093/nar/gku1078
Optimization of transcription factor binding map accuracy utilizing knockout-mouse models
Abstract
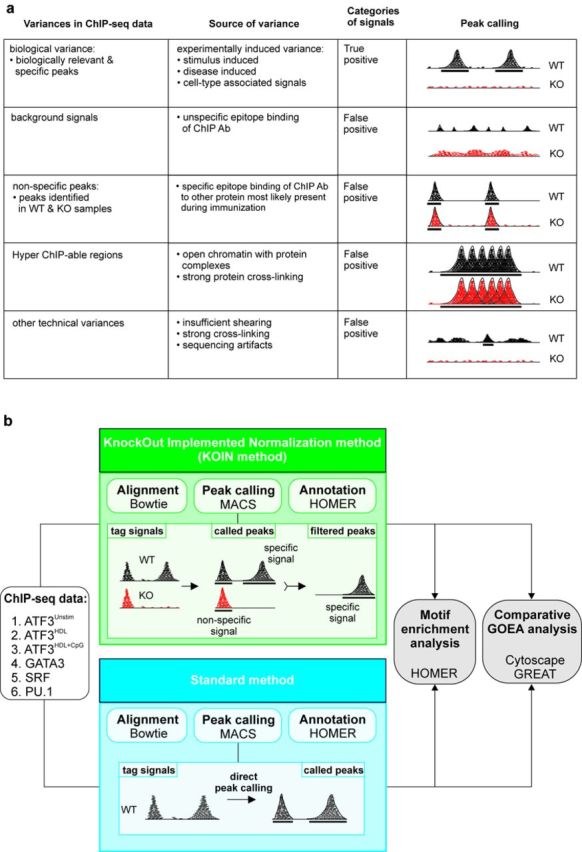
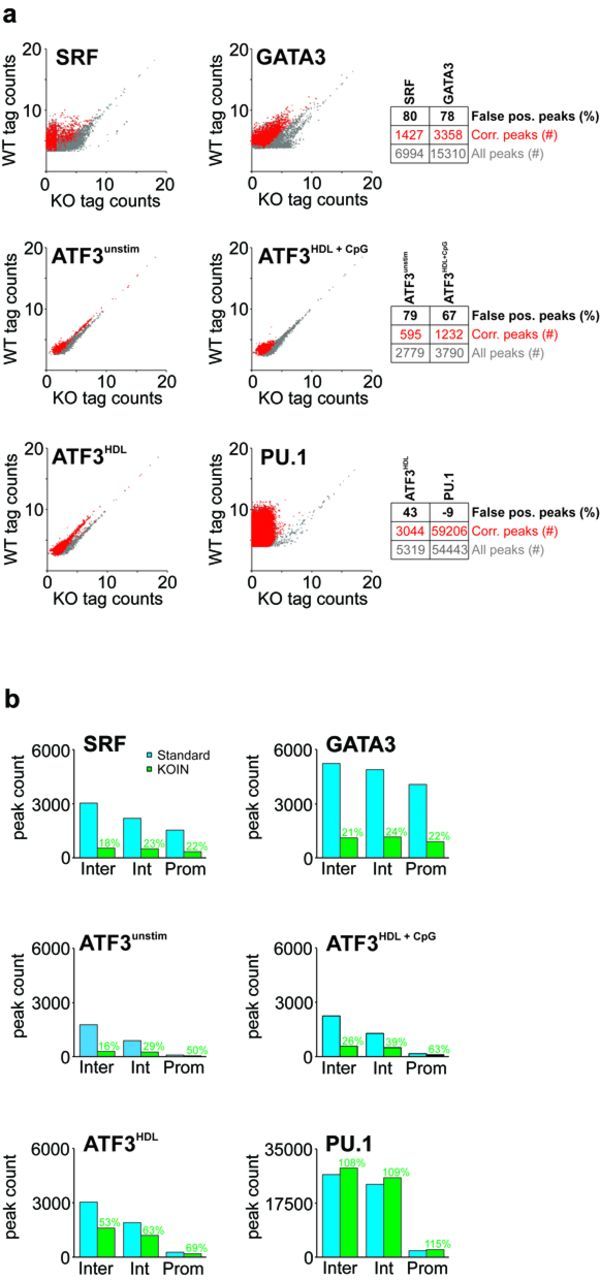
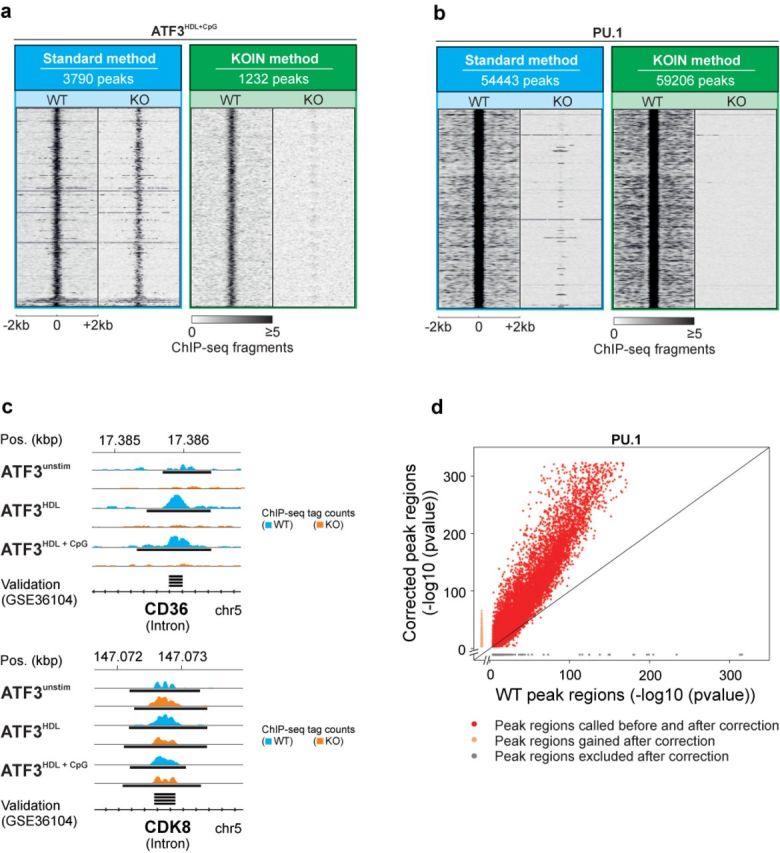
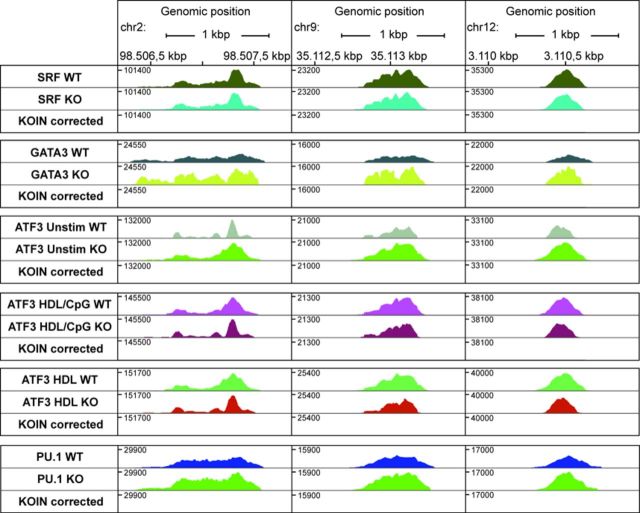
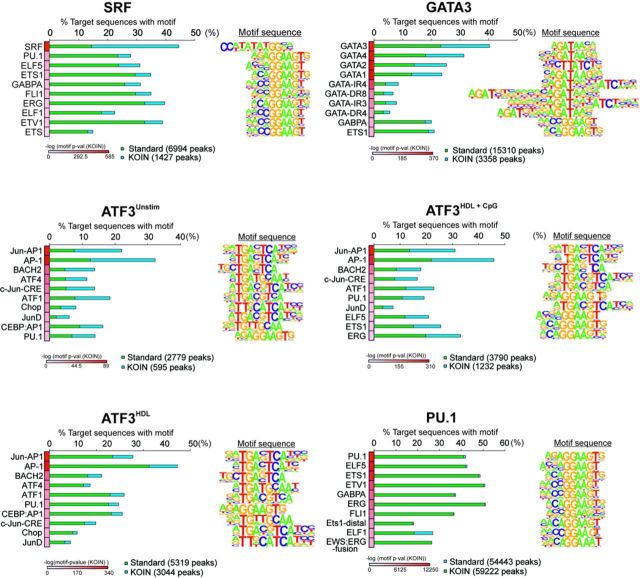
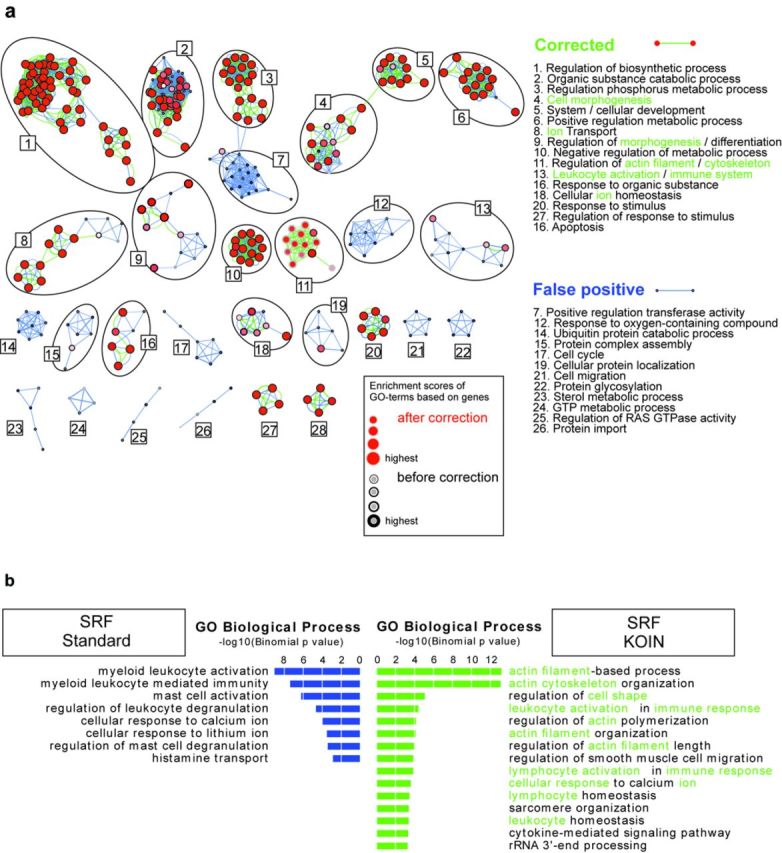
Genome-wide assessment of protein-DNA interaction by chromatin immunoprecipitation followed by massive parallel sequencing (ChIP-seq) is a key technology for studying transcription factor (TF) localization and regulation of gene expression. Signal-to-noise-ratio and signal specificity in ChIP-seq studies depend on many variables, including antibody affinity and specificity. Thus far, efforts to improve antibody reagents for ChIP-seq experiments have focused mainly on generating higher quality antibodies. Here we introduce KOIN (knockout implemented normalization) as a novel strategy to increase signal specificity and reduce noise by using TF knockout mice as a critical control for ChIP-seq data experiments. Additionally, KOIN can identify 'hyper ChIPable regions' as another source of false-positive signals. As the use of the KOIN algorithm reduces false-positive results and thereby prevents misinterpretation of ChIP-seq data, it should be considered as the gold standard for future ChIP-seq analyses, particularly when developing ChIP-assays with novel antibody reagents.
© The Author(s) 2014. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
