

Resolving the complexity of the human genome using single-molecule sequencing
- PMID: 25383537
- PMCID: PMC4317254
- DOI: 10.1038/nature13907
Resolving the complexity of the human genome using single-molecule sequencing
Abstract
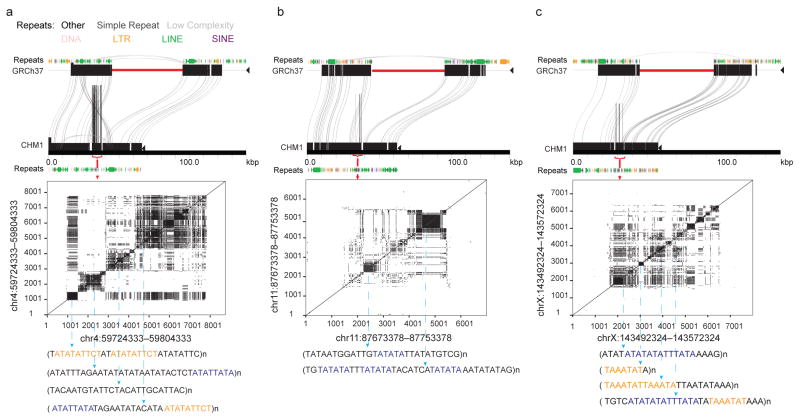
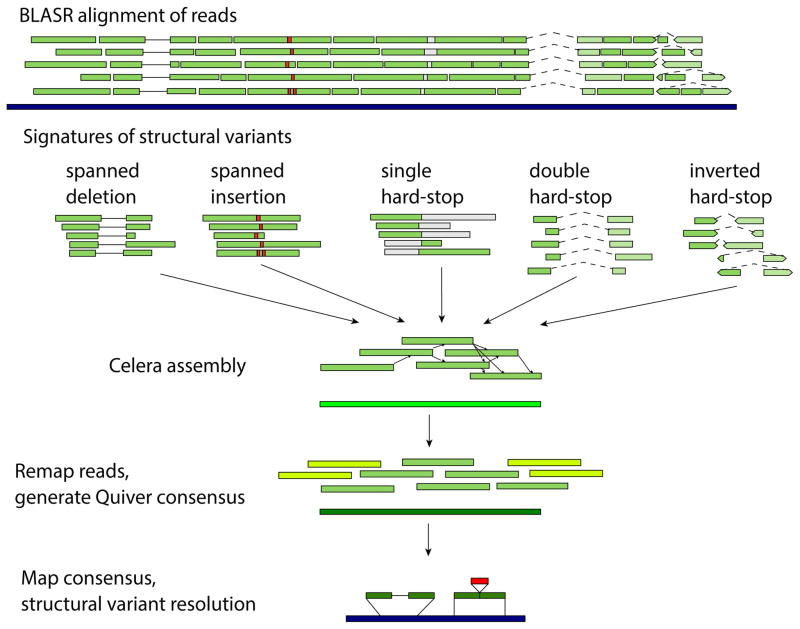
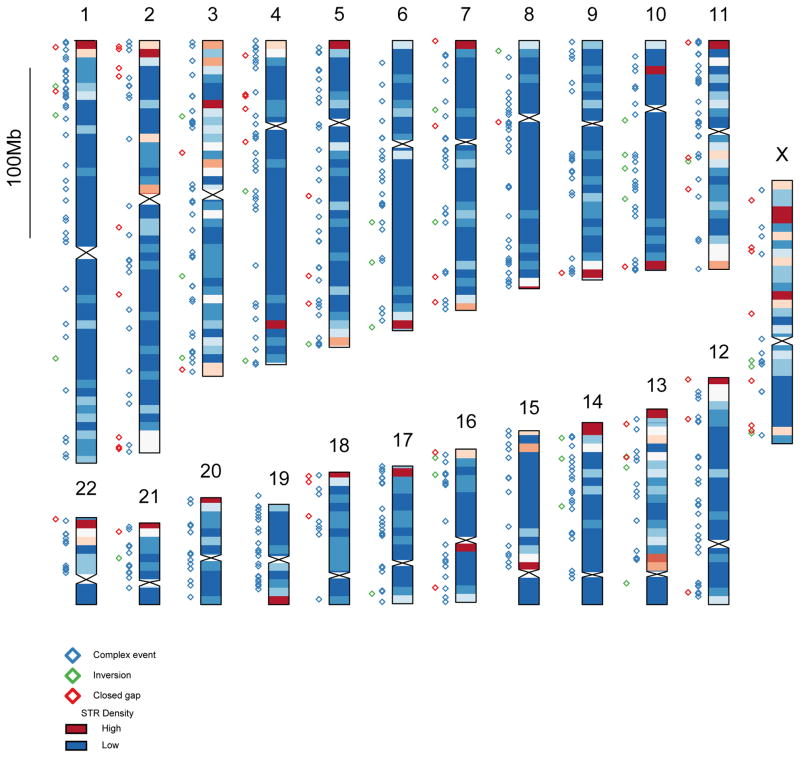
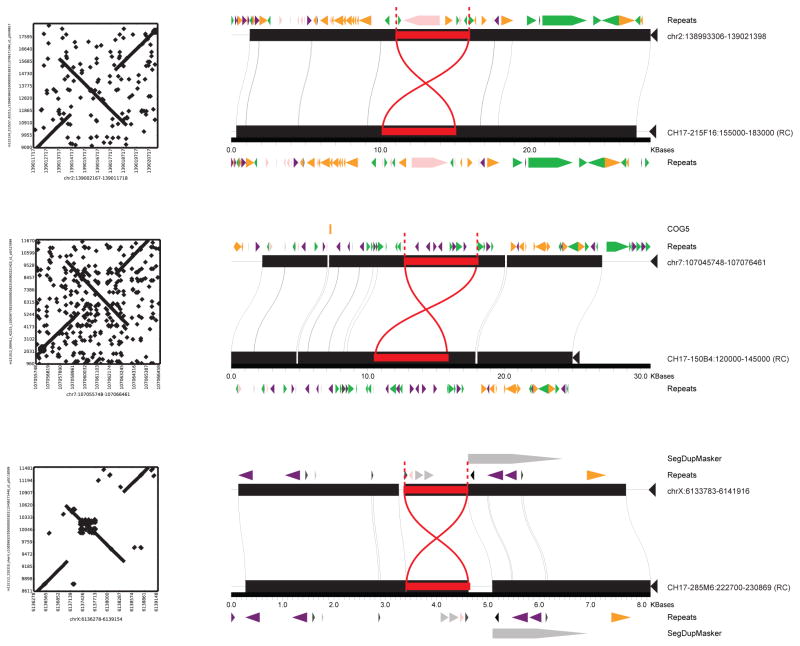
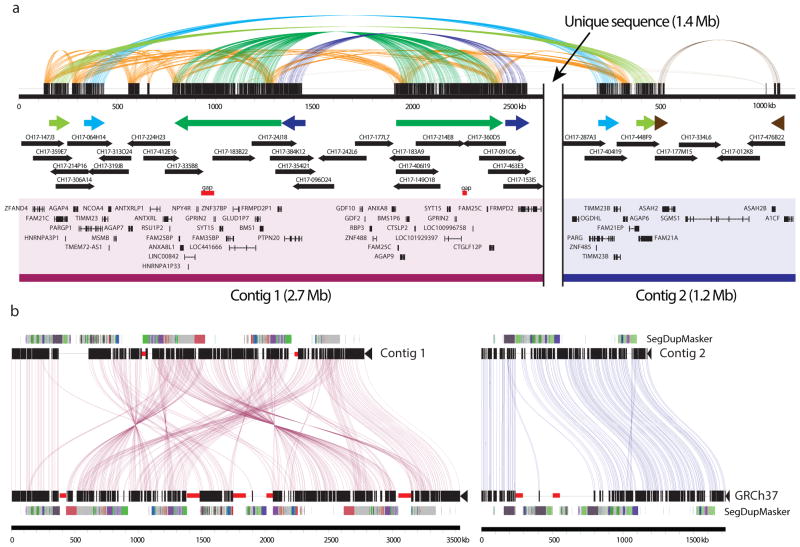
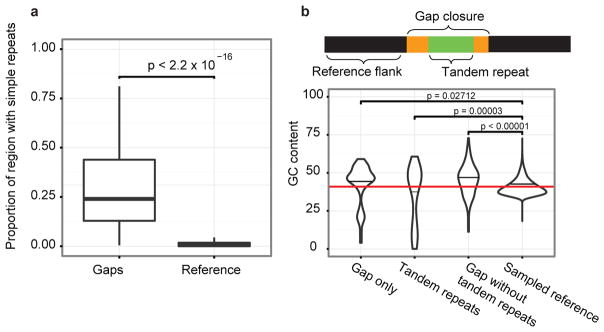
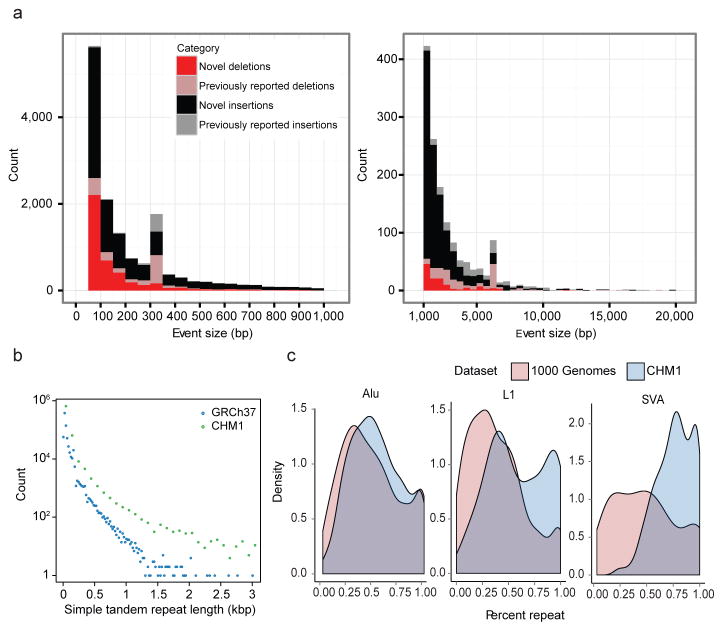
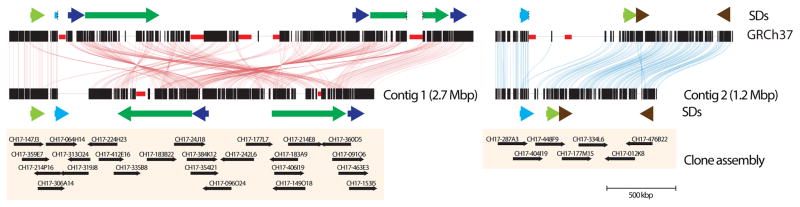
The human genome is arguably the most complete mammalian reference assembly, yet more than 160 euchromatic gaps remain and aspects of its structural variation remain poorly understood ten years after its completion. To identify missing sequence and genetic variation, here we sequence and analyse a haploid human genome (CHM1) using single-molecule, real-time DNA sequencing. We close or extend 55% of the remaining interstitial gaps in the human GRCh37 reference genome--78% of which carried long runs of degenerate short tandem repeats, often several kilobases in length, embedded within (G+C)-rich genomic regions. We resolve the complete sequence of 26,079 euchromatic structural variants at the base-pair level, including inversions, complex insertions and long tracts of tandem repeats. Most have not been previously reported, with the greatest increases in sensitivity occurring for events less than 5 kilobases in size. Compared to the human reference, we find a significant insertional bias (3:1) in regions corresponding to complex insertions and long short tandem repeats. Our results suggest a greater complexity of the human genome in the form of variation of longer and more complex repetitive DNA that can now be largely resolved with the application of this longer-read sequencing technology.
Conflict of interest statement
The authors declare competing financial interests. M.B., J.L., M.W.H. and J.K. are employees of Pacific Biosciences, Inc., a company commercializing DNA sequencing technologies; E.E.E. is on the scientific advisory board (SAB) of DNAnexus, Inc. and was formerly an SAB member of Pacific Biosciences, Inc. (2009–2013) and SynapDx Corp. (2011–2013); and M.J.C. was a former employee for Pacific Biosciences, Inc.
Figures

References
Publication types
MeSH terms
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
