

Analysis of nascent RNA identifies a unified architecture of initiation regions at mammalian promoters and enhancers
- PMID: 25383968
- PMCID: PMC4254663
- DOI: 10.1038/ng.3142
Analysis of nascent RNA identifies a unified architecture of initiation regions at mammalian promoters and enhancers
Abstract
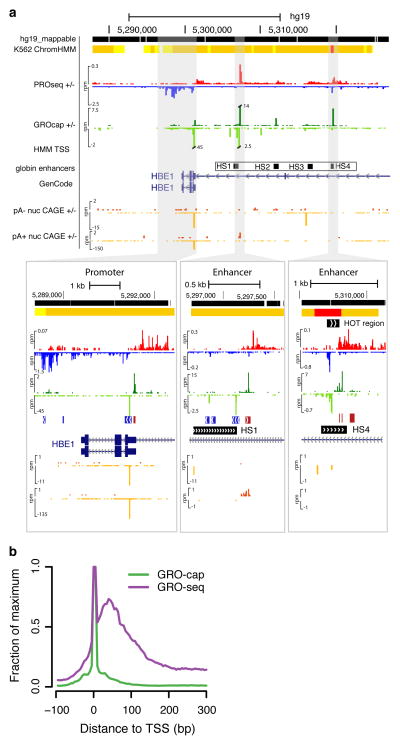
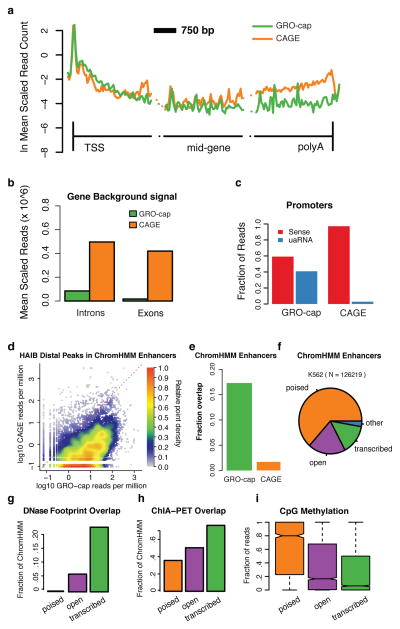
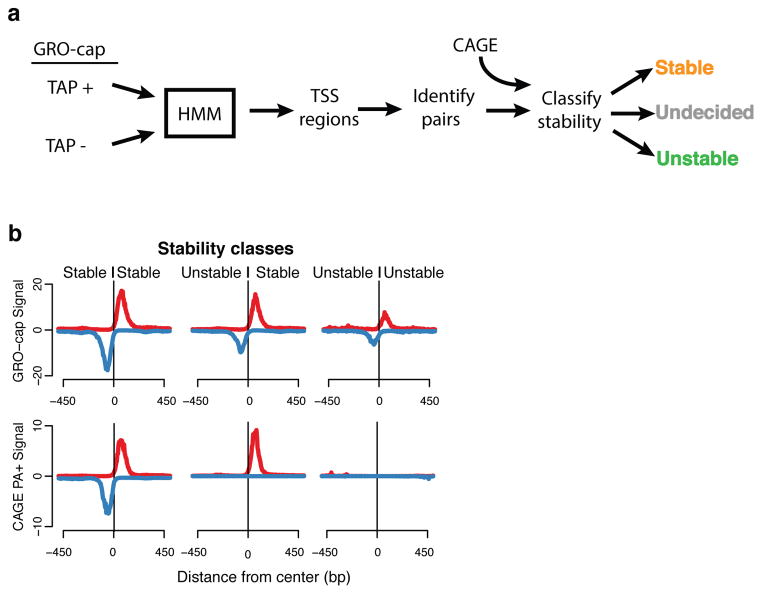
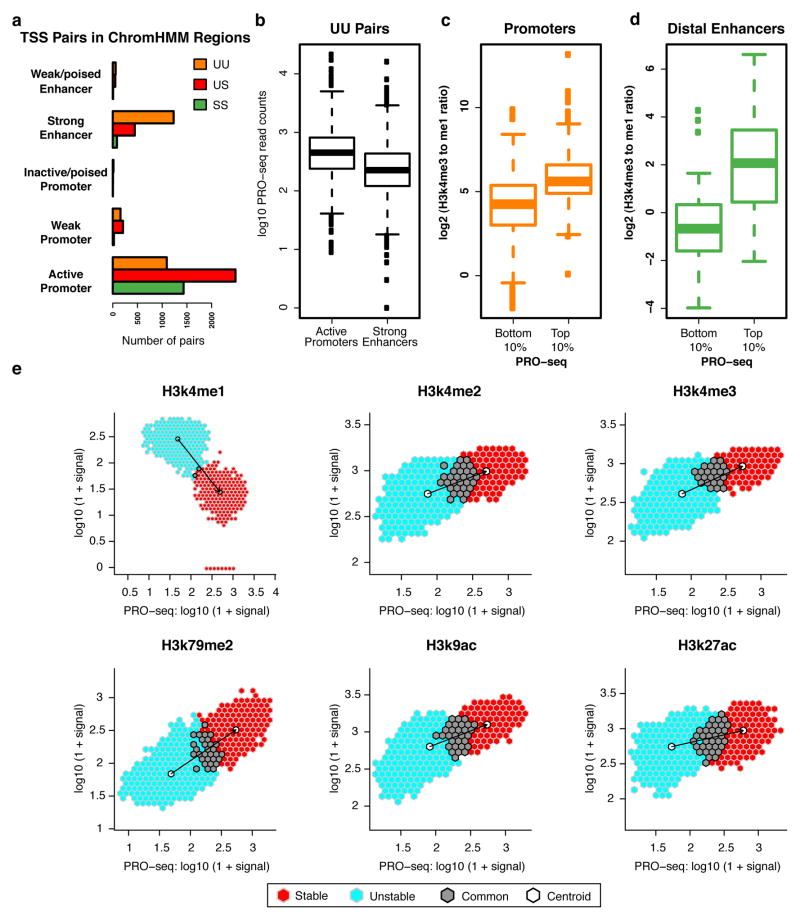
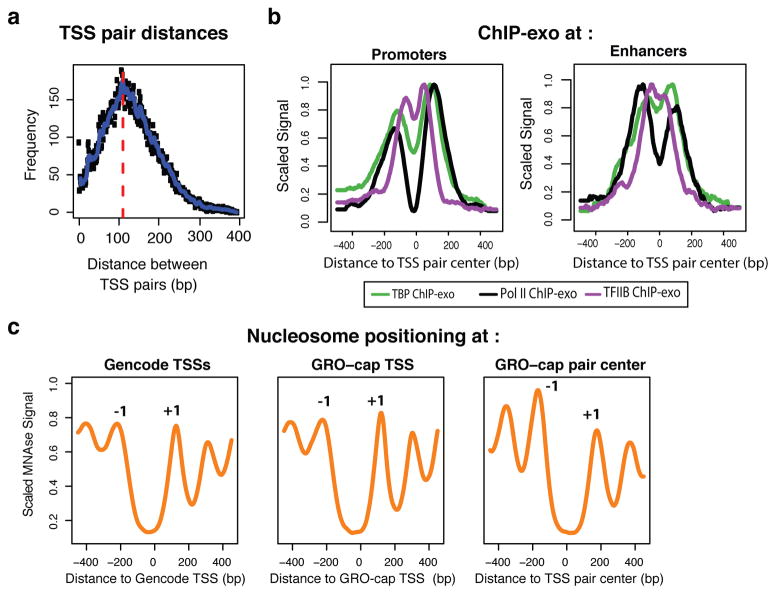
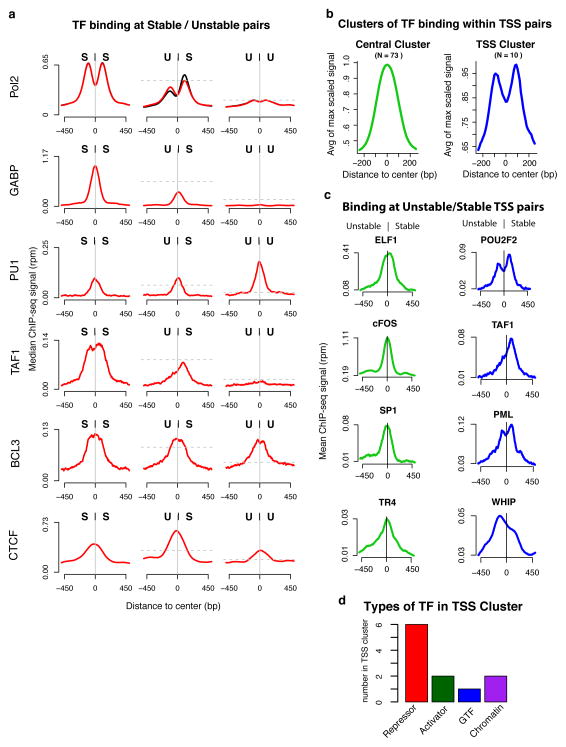
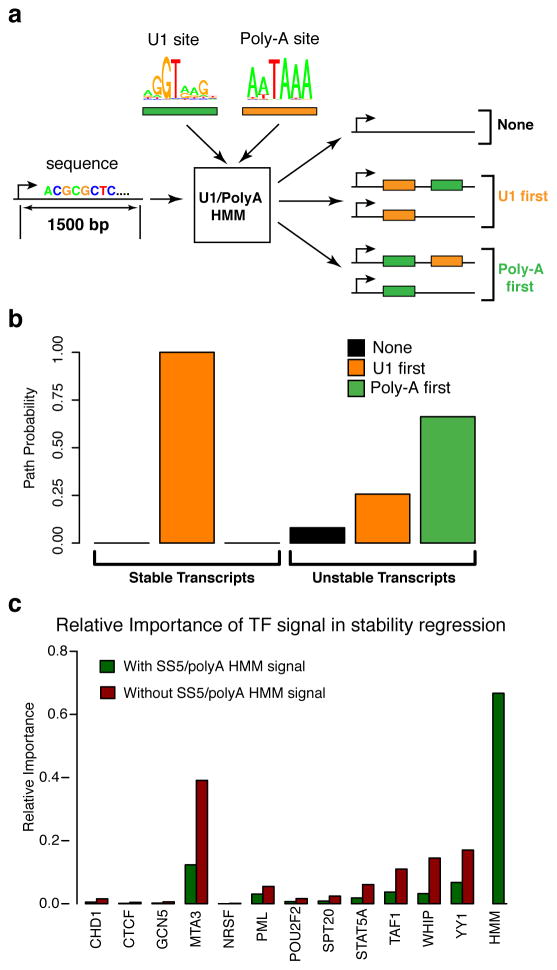
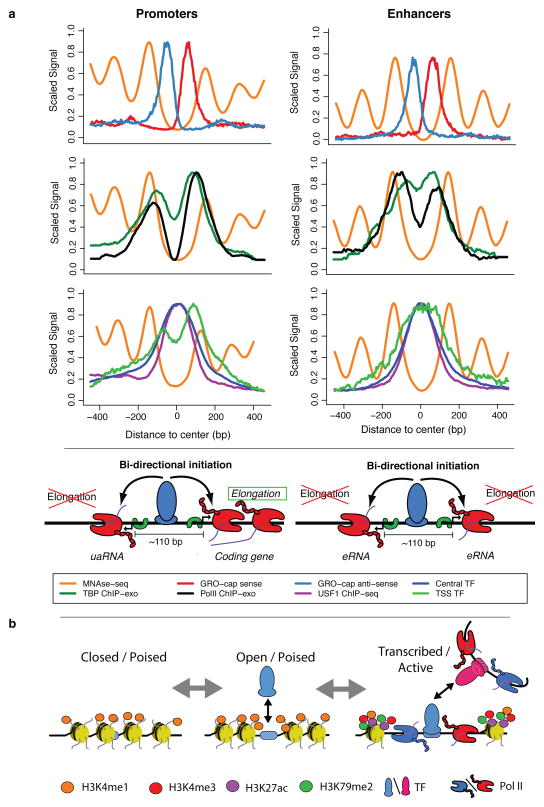
Despite the conventional distinction between them, promoters and enhancers share many features in mammals, including divergent transcription and similar modes of transcription factor binding. Here we examine the architecture of transcription initiation through comprehensive mapping of transcription start sites (TSSs) in human lymphoblastoid B cell (GM12878) and chronic myelogenous leukemic (K562) ENCODE Tier 1 cell lines. Using a nuclear run-on protocol called GRO-cap, which captures TSSs for both stable and unstable transcripts, we conduct detailed comparisons of thousands of promoters and enhancers in human cells. These analyses identify a common architecture of initiation, including tightly spaced (110 bp apart) divergent initiation, similar frequencies of core promoter sequence elements, highly positioned flanking nucleosomes and two modes of transcription factor binding. Post-initiation transcript stability provides a more fundamental distinction between promoters and enhancers than patterns of histone modification and association of transcription factors or co-activators. These results support a unified model of transcription initiation at promoters and enhancers.
Figures
Comment in
-
A shared architecture for promoters and enhancers.Nat Genet. 2014 Dec;46(12):1253-4. doi: 10.1038/ng.3152. Nat Genet. 2014. PMID: 25418744
References
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
