

Potential function for the Huntingtin protein as a scaffold for selective autophagy
- PMID: 25385587
- PMCID: PMC4250109
- DOI: 10.1073/pnas.1420103111
Potential function for the Huntingtin protein as a scaffold for selective autophagy
Abstract
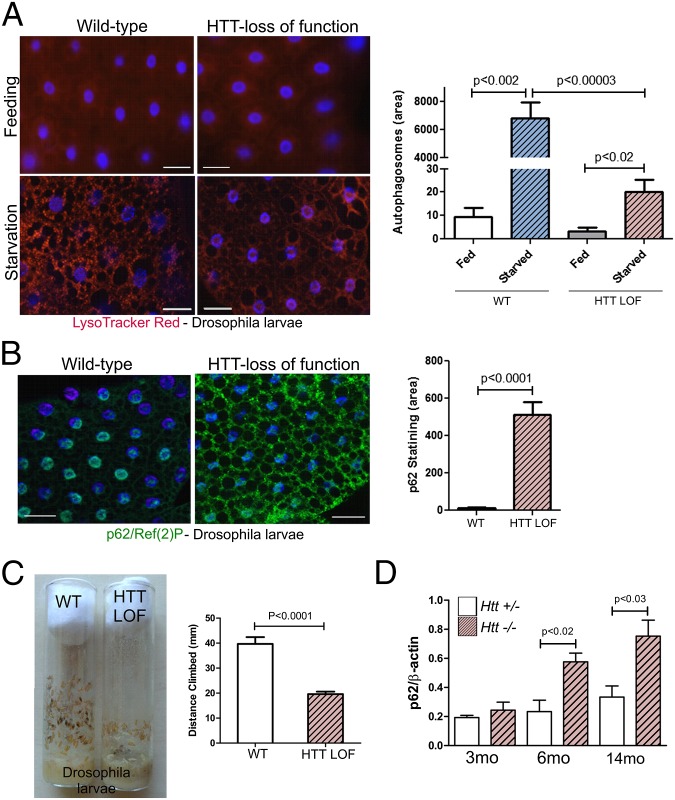
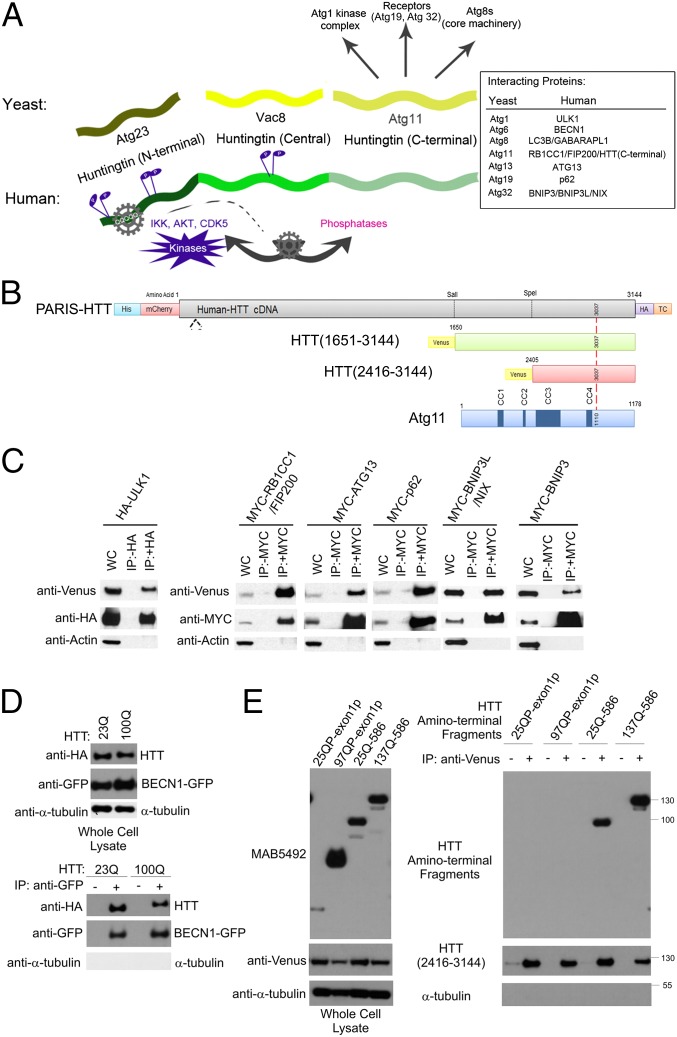
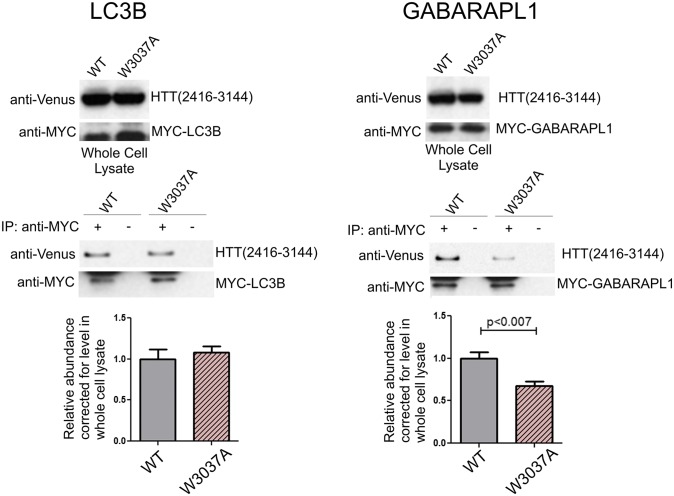
Although dominant gain-of-function triplet repeat expansions in the Huntingtin (HTT) gene are the underlying cause of Huntington disease (HD), understanding the normal functions of nonmutant HTT protein has remained a challenge. We report here findings that suggest that HTT plays a significant role in selective autophagy. Loss of HTT function in Drosophila disrupts starvation-induced autophagy in larvae and conditional knockout of HTT in the mouse CNS causes characteristic cellular hallmarks of disrupted autophagy, including an accumulation of striatal p62/SQSTM1 over time. We observe that specific domains of HTT have structural similarities to yeast Atg proteins that function in selective autophagy, and in particular that the C-terminal domain of HTT shares structural similarity to yeast Atg11, an autophagic scaffold protein. To explore possible functional similarity between HTT and Atg11, we investigated whether the C-terminal domain of HTT interacts with mammalian counterparts of yeast Atg11-interacting proteins. Strikingly, this domain of HTT coimmunoprecipitates with several key Atg11 interactors, including the Atg1/Unc-51-like autophagy activating kinase 1 kinase complex, autophagic receptor proteins, and mammalian Atg8 homologs. Mutation of a phylogenetically conserved WXXL domain in a C-terminal HTT fragment reduces coprecipitation with mammalian Atg8 homolog GABARAPL1, suggesting a direct interaction. Collectively, these data support a possible central role for HTT as an Atg11-like scaffold protein. These findings have relevance to both mechanisms of disease pathogenesis and to therapeutic intervention strategies that reduce levels of both mutant and normal HTT.
Keywords: Huntingtin; Huntington disease; neurodegeneration; polyglutamine; selective autophagy.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Ross CA, Tabrizi SJ. Huntington’s disease: From molecular pathogenesis to clinical treatment. Lancet Neurol. 2011;10(1):83–98. - PubMed
-
- Duyao MP, et al. Inactivation of the mouse Huntington’s disease gene homolog Hdh. Science. 1995;269(5222):407–410. - PubMed
-
- Zeitlin S, Liu JP, Chapman DL, Papaioannou VE, Efstratiadis A. Increased apoptosis and early embryonic lethality in mice nullizygous for the Huntington’s disease gene homologue. Nat Genet. 1995;11(2):155–163. - PubMed
-
- Nasir J, et al. Targeted disruption of the Huntington’s disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell. 1995;81(5):811–823. - PubMed
-
- Dragatsis I, Levine MS, Zeitlin S. Inactivation of Hdh in the brain and testis results in progressive neurodegeneration and sterility in mice. Nat Genet. 2000;26(3):300–306. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
