

The role of endoplasmic reticulum stress in human pathology
- PMID: 25387057
- PMCID: PMC5568783
- DOI: 10.1146/annurev-pathol-012513-104649
The role of endoplasmic reticulum stress in human pathology
Abstract
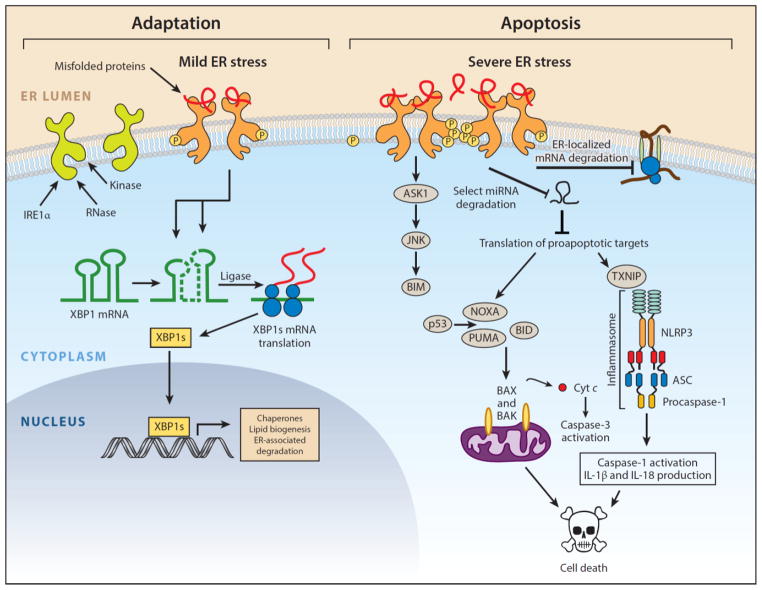
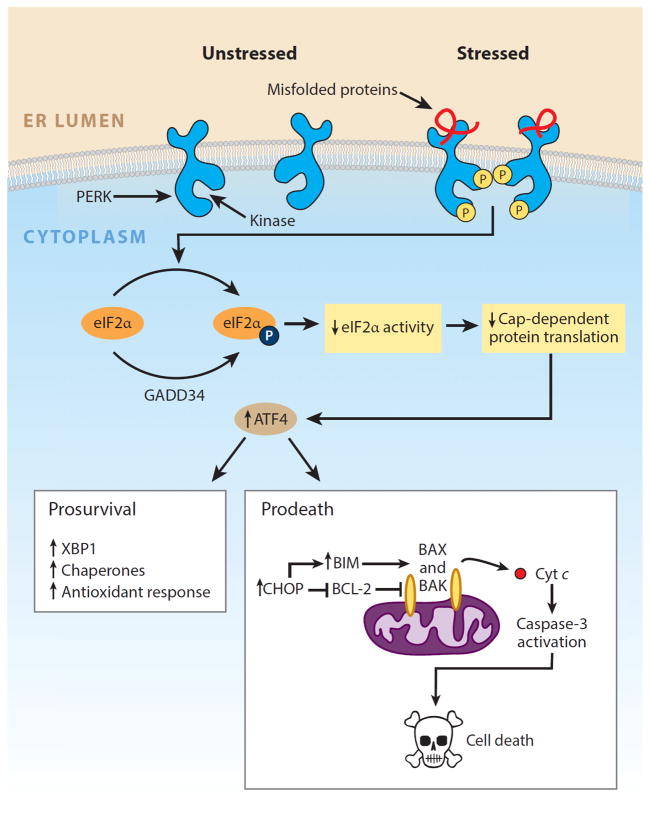
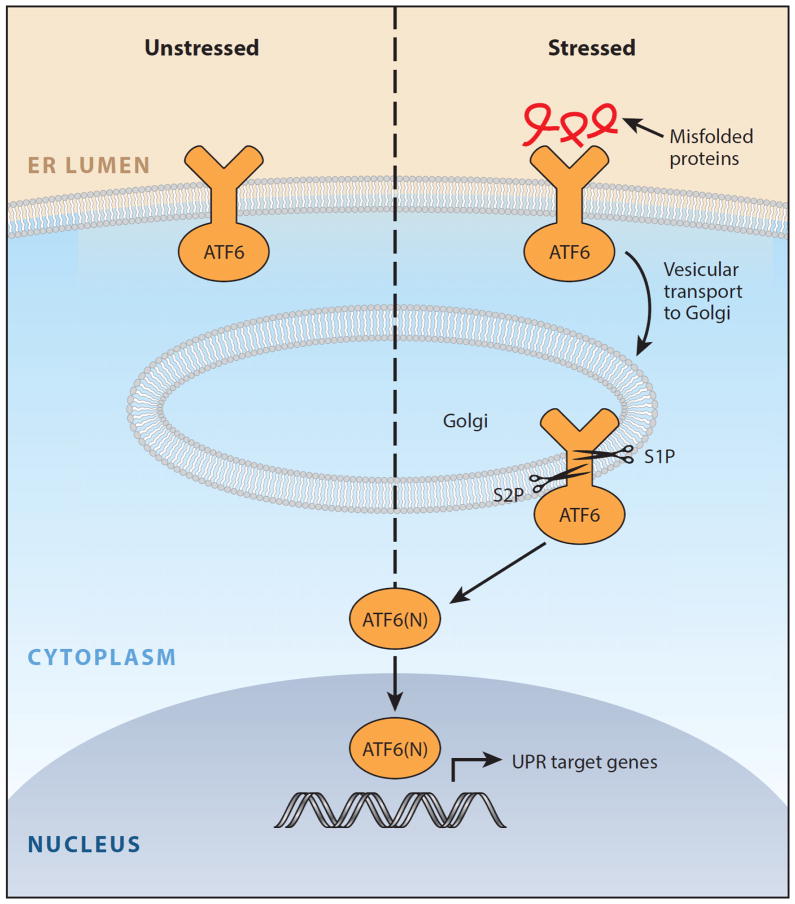
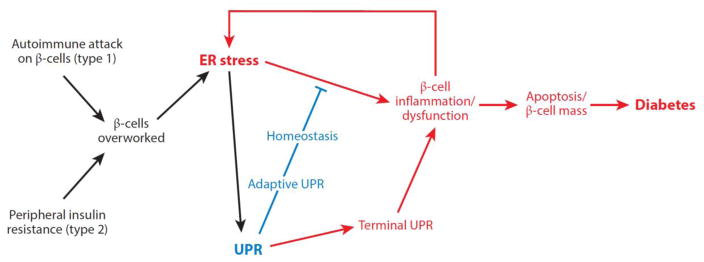
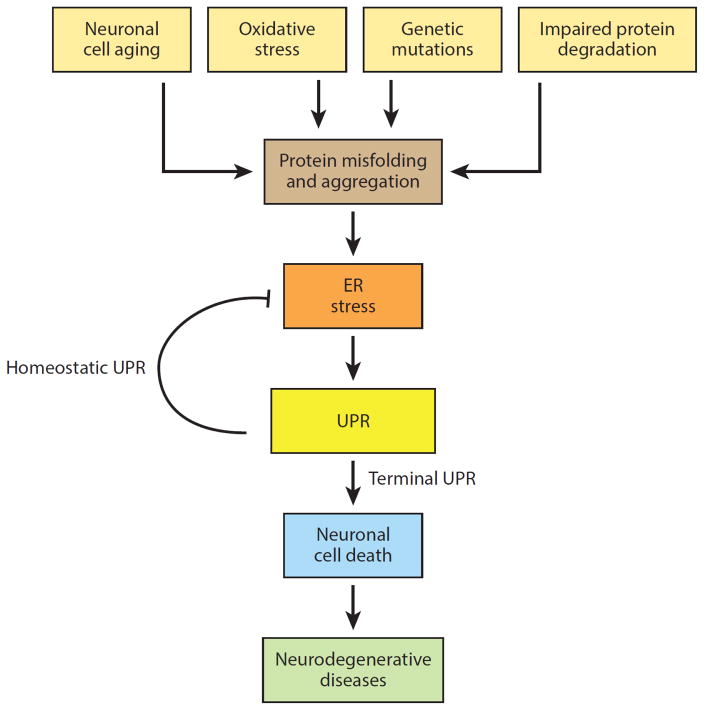
Numerous genetic and environmental insults impede the ability of cells to properly fold and posttranslationally modify secretory and transmembrane proteins in the endoplasmic reticulum (ER), leading to a buildup of misfolded proteins in this organelle--a condition called ER stress. ER-stressed cells must rapidly restore protein-folding capacity to match protein-folding demand if they are to survive. In the presence of high levels of misfolded proteins in the ER, an intracellular signaling pathway called the unfolded protein response (UPR) induces a set of transcriptional and translational events that restore ER homeostasis. However, if ER stress persists chronically at high levels, a terminal UPR program ensures that cells commit to self-destruction. Chronic ER stress and defects in UPR signaling are emerging as key contributors to a growing list of human diseases, including diabetes, neurodegeneration, and cancer. Hence, there is much interest in targeting components of the UPR as a therapeutic strategy to combat these ER stress-associated pathologies.
Keywords: apoptosis; cancer; diabetes; neurodegeneration; protein misfolding; unfolded protein response.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
