

Further improvements to linear mixed models for genome-wide association studies
- PMID: 25387525
- PMCID: PMC4230738
- DOI: 10.1038/srep06874
Further improvements to linear mixed models for genome-wide association studies
Abstract
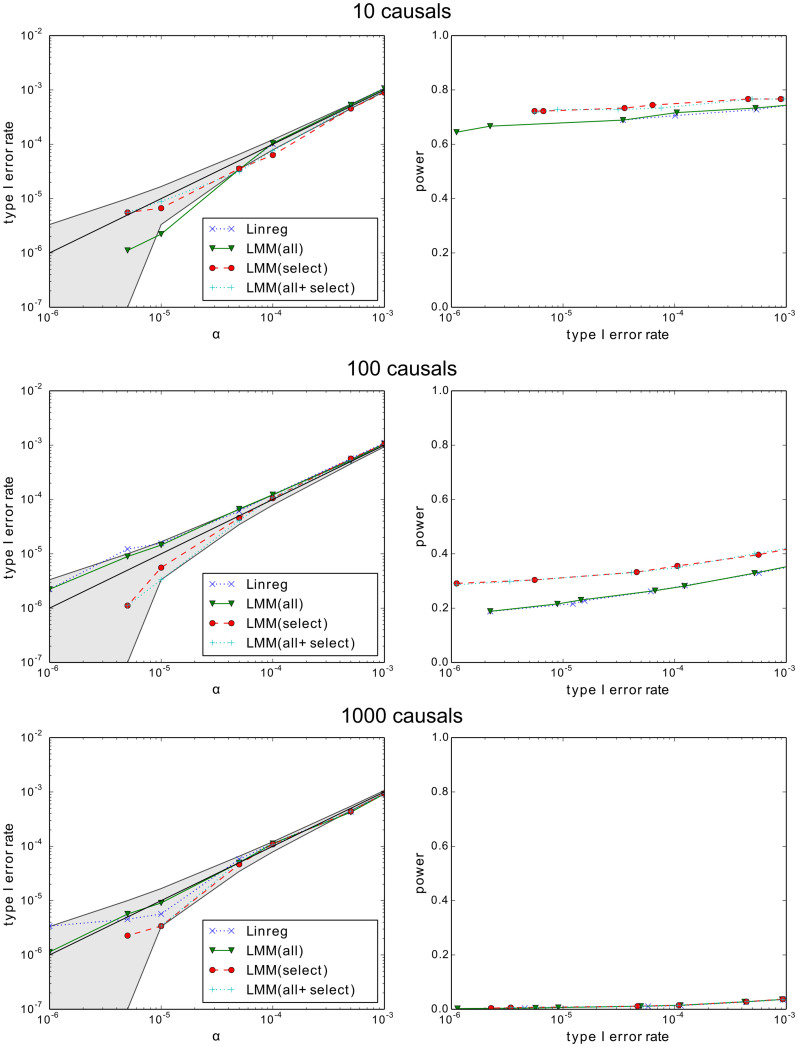
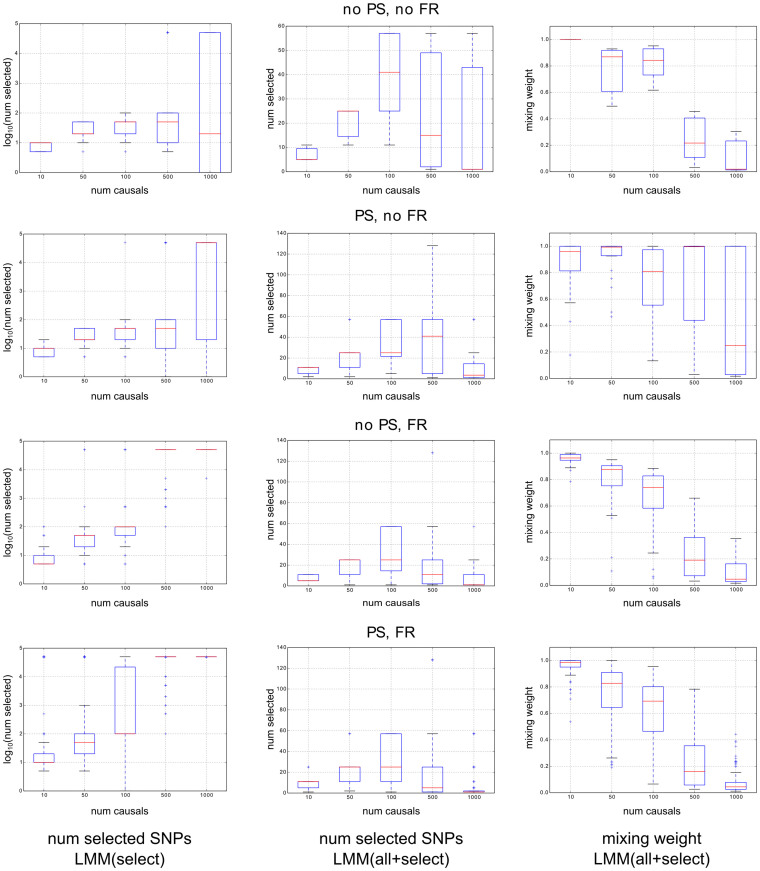
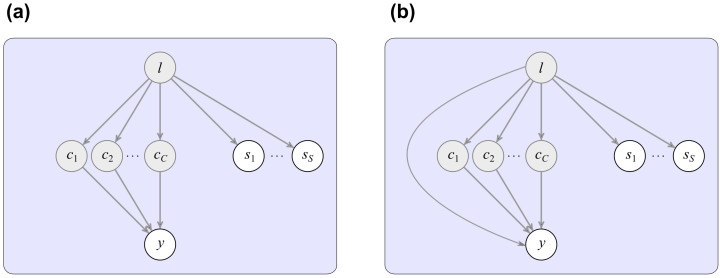
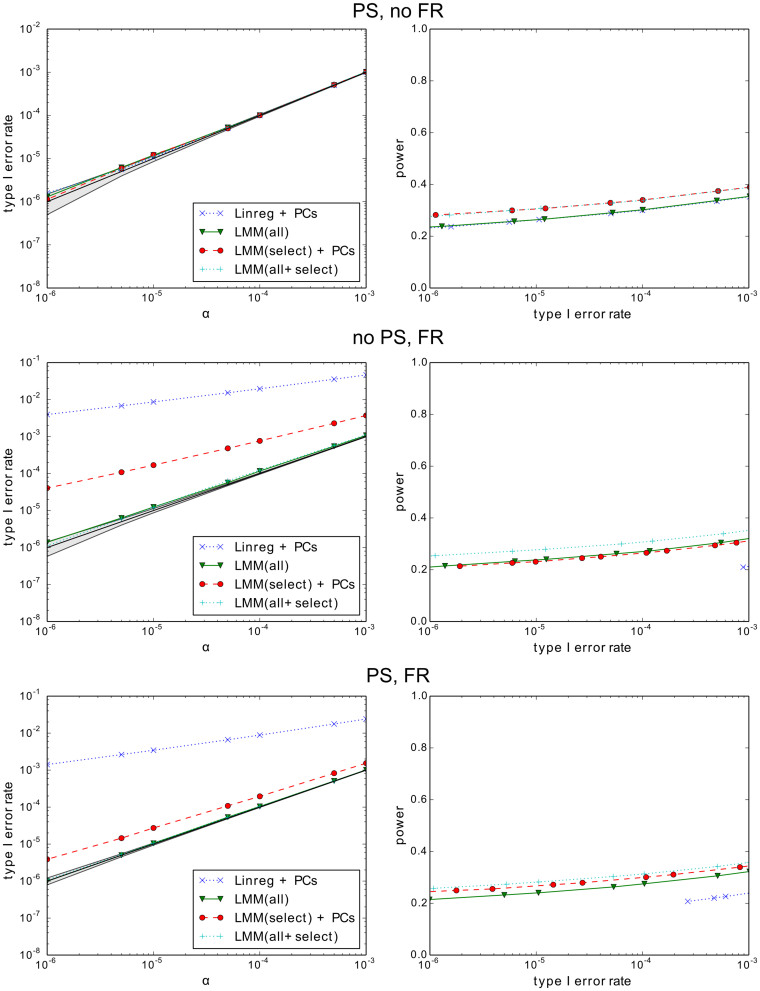
We examine improvements to the linear mixed model (LMM) that better correct for population structure and family relatedness in genome-wide association studies (GWAS). LMMs rely on the estimation of a genetic similarity matrix (GSM), which encodes the pairwise similarity between every two individuals in a cohort. These similarities are estimated from single nucleotide polymorphisms (SNPs) or other genetic variants. Traditionally, all available SNPs are used to estimate the GSM. In empirical studies across a wide range of synthetic and real data, we find that modifications to this approach improve GWAS performance as measured by type I error control and power. Specifically, when only population structure is present, a GSM constructed from SNPs that well predict the phenotype in combination with principal components as covariates controls type I error and yields more power than the traditional LMM. In any setting, with or without population structure or family relatedness, a GSM consisting of a mixture of two component GSMs, one constructed from all SNPs and another constructed from SNPs that well predict the phenotype again controls type I error and yields more power than the traditional LMM. Software implementing these improvements and the experimental comparisons are available at http://microsoft.com/science.
Conflict of interest statement
C.W., C.L., N.F., C.K., R.D., J.L., and D.H. were employed by Microsoft while performing this work.
Figures
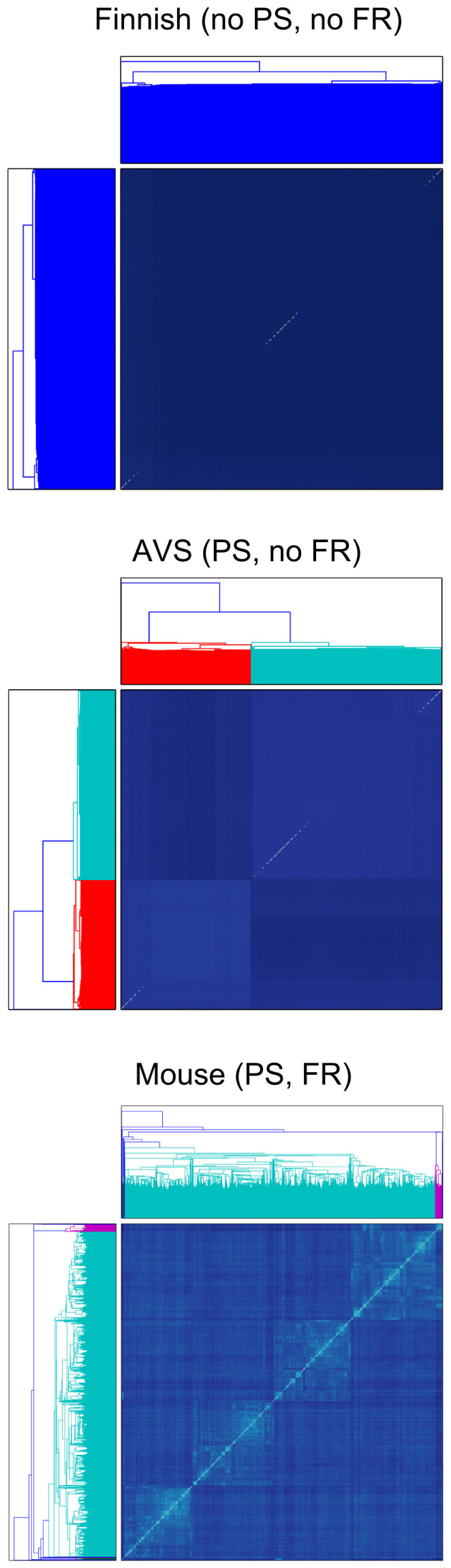
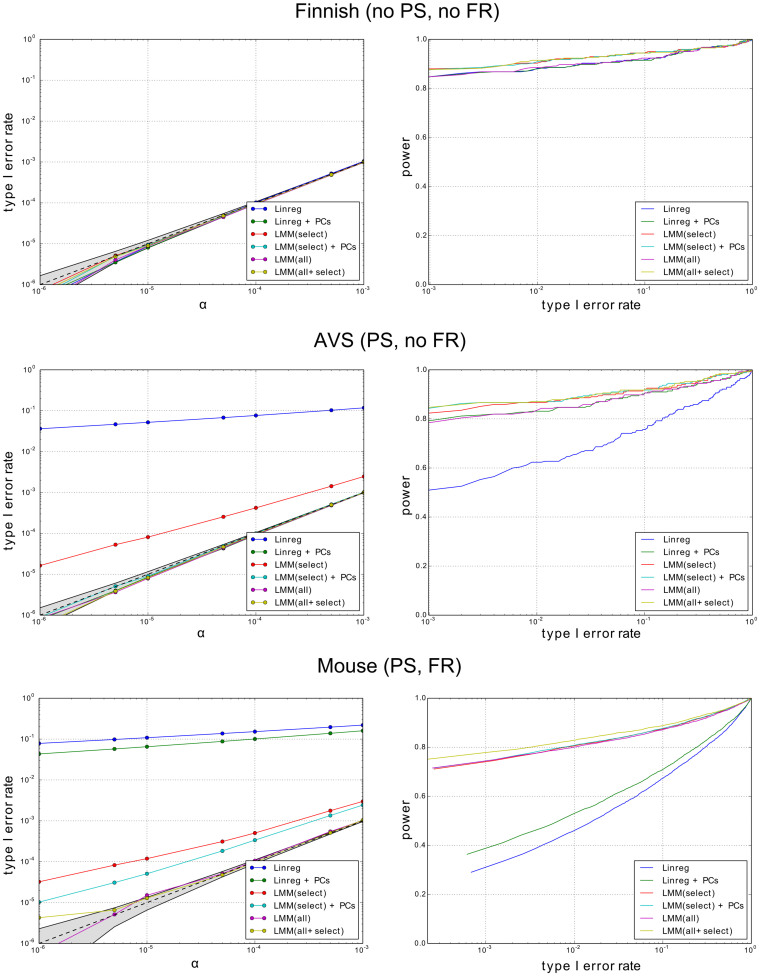
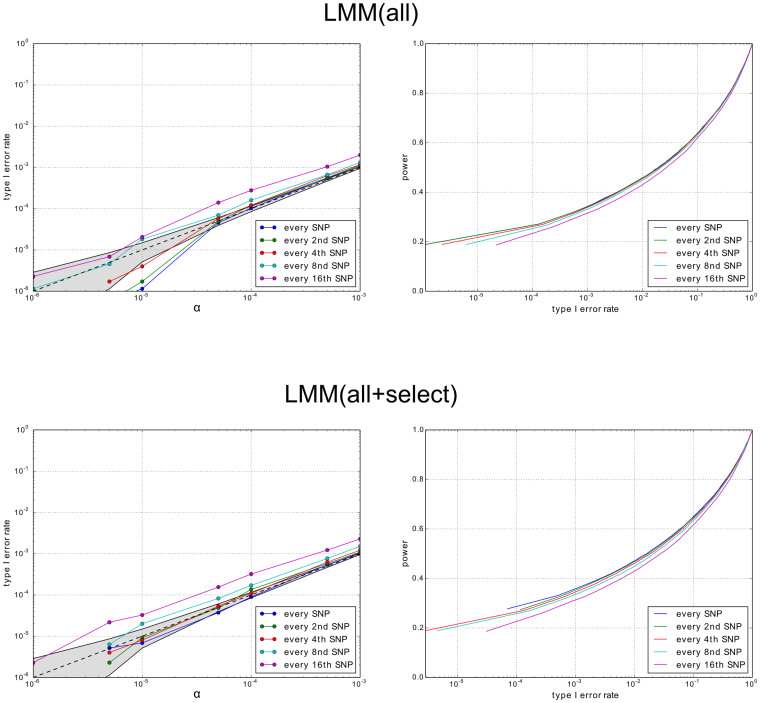
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
