

Whole-genome enrichment and sequencing of Chlamydia trachomatis directly from clinical samples
- PMID: 25388670
- PMCID: PMC4233057
- DOI: 10.1186/s12879-014-0591-3
Whole-genome enrichment and sequencing of Chlamydia trachomatis directly from clinical samples
Abstract
Background: Chlamydia trachomatis is a pathogen of worldwide importance, causing more than 100 million cases of sexually transmitted infections annually. Whole-genome sequencing is a powerful high resolution tool that can be used to generate accurate data on bacterial population structure, phylogeography and mutations associated with antimicrobial resistance. The objective of this study was to perform whole-genome enrichment and sequencing of C. trachomatis directly from clinical samples.
Methods: C. trachomatis positive samples comprising seven vaginal swabs and three urine samples were sequenced without prior in vitro culture in addition to nine cultured C. trachomatis samples, representing different serovars. A custom capture RNA bait set, that captures all known diversity amongst C. trachomatis genomes, was used in a whole-genome enrichment step during library preparation to enrich for C. trachomatis DNA. All samples were sequenced on the MiSeq platform.
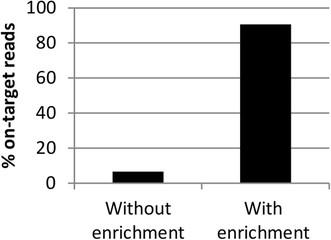
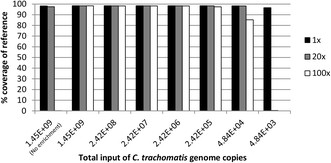
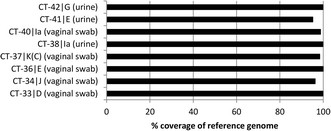
Results: Full length C. trachomatis genomes (>95-100% coverage of a reference genome) were successfully generated for eight of ten clinical samples and for all cultured samples. The proportion of reads mapping to C. trachomatis and the mean read depth across each genome were strongly linked to the number of bacterial copies within the original sample. Phylogenetic analysis confirmed the known population structure and the data showed potential for identification of minority variants and mutations associated with antimicrobial resistance. The sensitivity of the method was >10-fold higher than other reported methodologies.
Conclusions: The combination of whole-genome enrichment and deep sequencing has proven to be a non-mutagenic approach, capturing all known variation found within C. trachomatis genomes. The method is a consistent and sensitive tool that enables rapid whole-genome sequencing of C. trachomatis directly from clinical samples and has the potential to be adapted to other pathogens with a similar clonal nature.
Figures



Similar articles
-
Whole-Genome Enrichment Using RNA Probes and Sequencing of Chlamydia trachomatis Directly from Clinical Samples.Methods Mol Biol. 2017;1616:1-22. doi: 10.1007/978-1-4939-7037-7_1. Methods Mol Biol. 2017. PMID: 28600759
-
Whole-Genome Sequencing of Chlamydia trachomatis Directly from Human Samples.Methods Mol Biol. 2019;2042:45-67. doi: 10.1007/978-1-4939-9694-0_6. Methods Mol Biol. 2019. PMID: 31385270
-
Whole-Genome Enrichment and Sequencing of Chlamydia trachomatis Directly from Patient Clinical Vaginal and Rectal Swabs.mSphere. 2021 Mar 3;6(2):e01302-20. doi: 10.1128/mSphere.01302-20. mSphere. 2021. PMID: 33658279 Free PMC article.
-
Evolution of Chlamydia trachomatis.Ann N Y Acad Sci. 2011 Aug;1230:E11-8. doi: 10.1111/j.1749-6632.2011.06194.x. Ann N Y Acad Sci. 2011. PMID: 22239534 Review.
-
High-resolution typing of Chlamydia trachomatis: epidemiological and clinical uses.Curr Opin Infect Dis. 2015 Feb;28(1):61-71. doi: 10.1097/QCO.0000000000000129. Curr Opin Infect Dis. 2015. PMID: 25490105 Review.
Cited by
-
Host-Associated Genomic Features of the Novel Uncultured Intracellular Pathogen Ca. Ichthyocystis Revealed by Direct Sequencing of Epitheliocysts.Genome Biol Evol. 2016 Jun 13;8(6):1672-89. doi: 10.1093/gbe/evw111. Genome Biol Evol. 2016. PMID: 27190004 Free PMC article.
-
Resistome and virulome determination in Helicobacter pylori using next-generation sequencing with target-enrichment technology.Microbiol Spectr. 2025 Apr;13(4):e0329824. doi: 10.1128/spectrum.03298-24. Epub 2025 Mar 5. Microbiol Spectr. 2025. PMID: 40042287 Free PMC article.
-
Evolutionary dynamics in the genome of ocular Chlamydia trachomatis strains from Northern Tanzania following mass drug administration.Microb Genom. 2025 Jul;11(7):001431. doi: 10.1099/mgen.0.001431. Microb Genom. 2025. PMID: 40601472 Free PMC article.
-
Molecular Capture of Mycobacterium tuberculosis Genomes Directly from Clinical Samples: A Potential Backup Approach for Epidemiological and Drug Susceptibility Inferences.Int J Mol Sci. 2023 Feb 2;24(3):2912. doi: 10.3390/ijms24032912. Int J Mol Sci. 2023. PMID: 36769230 Free PMC article.
-
Chlamydia trachomatis Biovar L2 Infection in Women in South Africa.Emerg Infect Dis. 2017 Nov;23(11):1913-1915. doi: 10.3201/eid2311.170758. Emerg Infect Dis. 2017. PMID: 29048296 Free PMC article.
References
-
- WHO. 2011: Prevalence and Incidence of Selected Sexually Transmitted Infections, Chlamydia Trachomatis, Neisseria Gonorrhoeae, Syphilis and Trichomonas Vaginalis: Methods an Results Used by WHO to Generate 2005 Estimate.: World Health Organization; 2011:1-38.
-
- WHO. 2012: Global Incidence and Prevalence of Selected Curable Sexually Transmitted Infections - 2008.: World Health Organization; 2012:1-28.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
