

Mechismo: predicting the mechanistic impact of mutations and modifications on molecular interactions
- PMID: 25392414
- PMCID: PMC4333368
- DOI: 10.1093/nar/gku1094
Mechismo: predicting the mechanistic impact of mutations and modifications on molecular interactions
Abstract
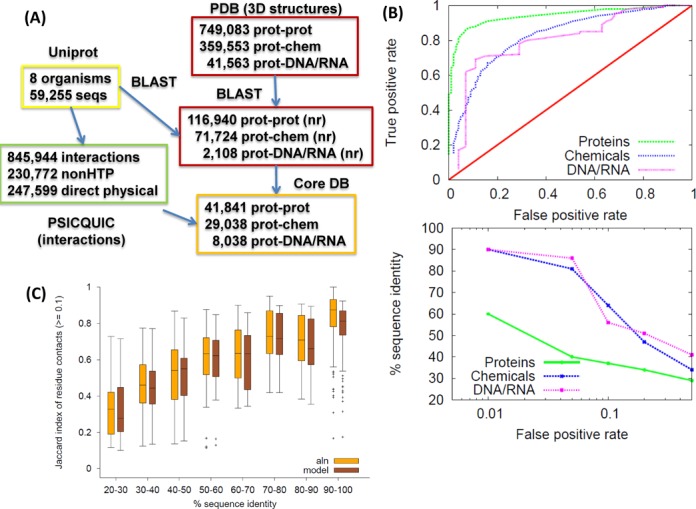
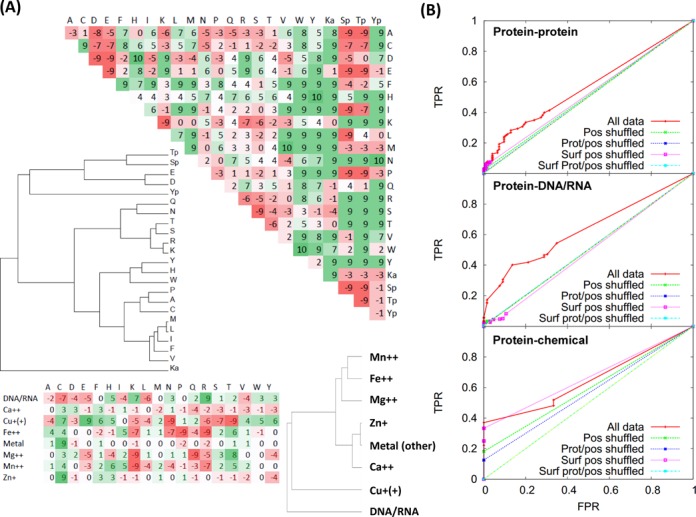
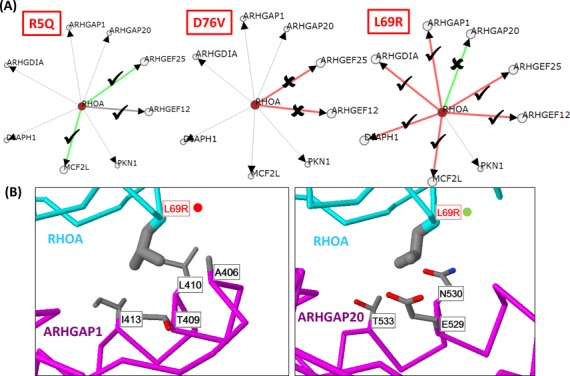
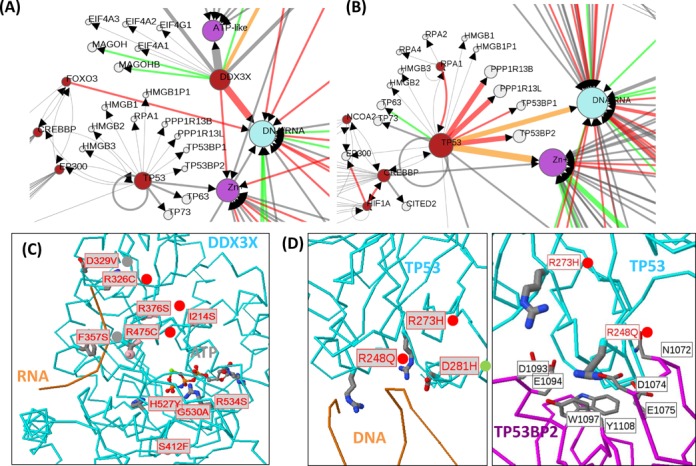
Systematic interrogation of mutation or protein modification data is important to identify sites with functional consequences and to deduce global consequences from large data sets. Mechismo (mechismo.russellab.org) enables simultaneous consideration of thousands of 3D structures and biomolecular interactions to predict rapidly mechanistic consequences for mutations and modifications. As useful functional information often only comes from homologous proteins, we benchmarked the accuracy of predictions as a function of protein/structure sequence similarity, which permits the use of relatively weak sequence similarities with an appropriate confidence measure. For protein-protein, protein-nucleic acid and a subset of protein-chemical interactions, we also developed and benchmarked a measure of whether modifications are likely to enhance or diminish the interactions, which can assist the detection of modifications with specific effects. Analysis of high-throughput sequencing data shows that the approach can identify interesting differences between cancers, and application to proteomics data finds potential mechanistic insights for how post-translational modifications can alter biomolecular interactions.
© The Author(s) 2014. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
-
- Kilpivaara O., Aaltonen L.A. Diagnostic cancer genome sequencing and the contribution of germline variants. Science. 2013;339:1559–1562. - PubMed
-
- Choudhary C., Mann M. Decoding signalling networks by mass spectrometry-based proteomics. Nat. Rev. Mol. Cell Biol. 2010;11:427–439. - PubMed
-
- Mosca R., Céol A., Aloy P. Interactome3D: adding structural details to protein networks. Nat. Methods. 2013;10:47–53. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
