

Evolution of DNA methylation patterns in the Brassicaceae is driven by differences in genome organization
- PMID: 25393550
- PMCID: PMC4230842
- DOI: 10.1371/journal.pgen.1004785
Evolution of DNA methylation patterns in the Brassicaceae is driven by differences in genome organization
Abstract
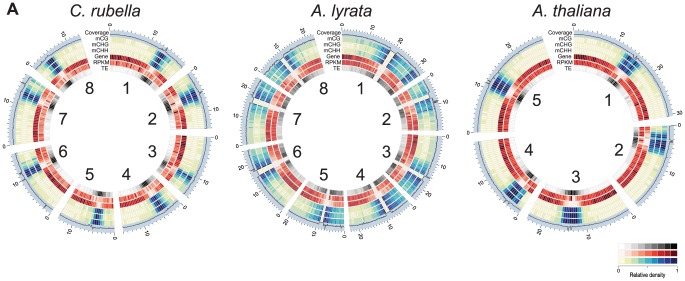
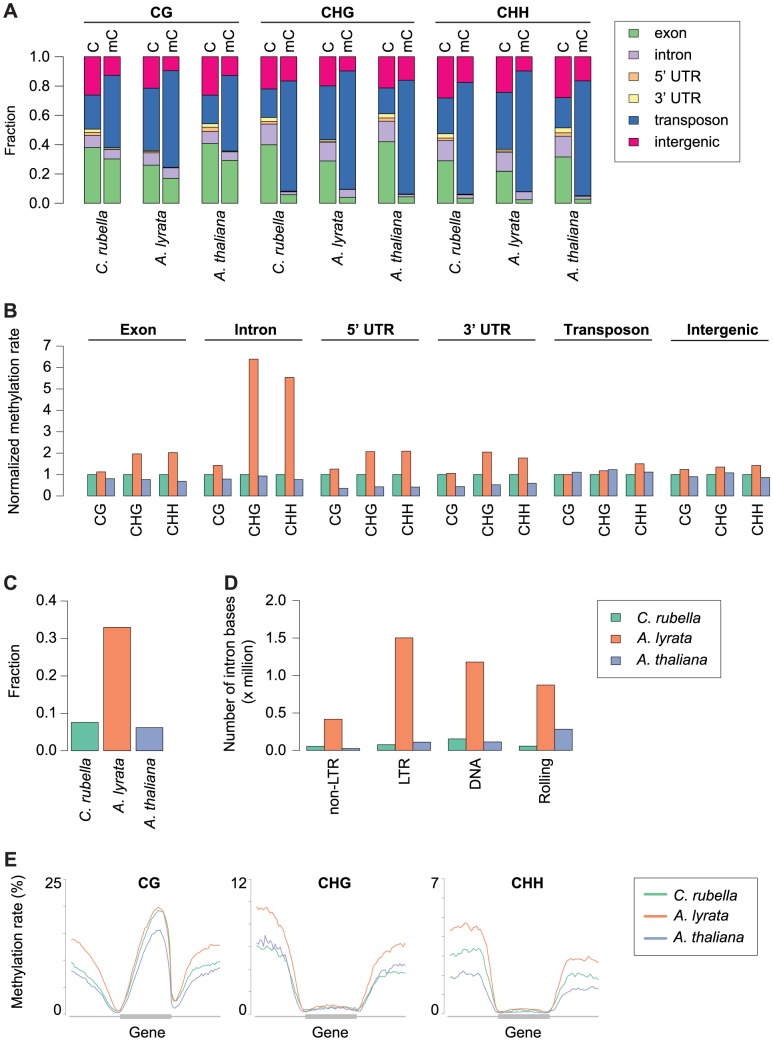
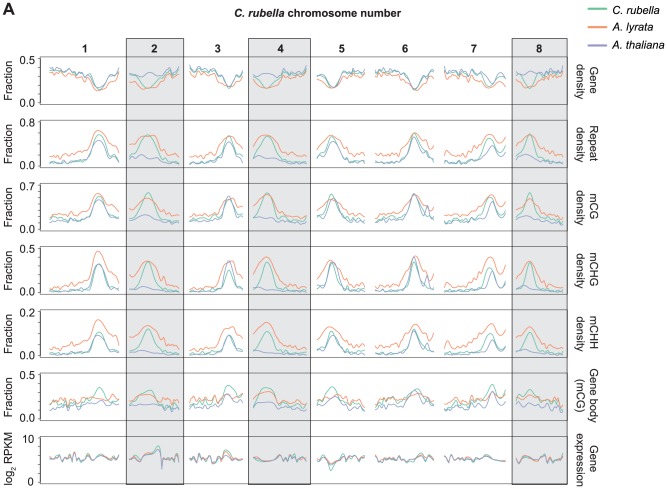
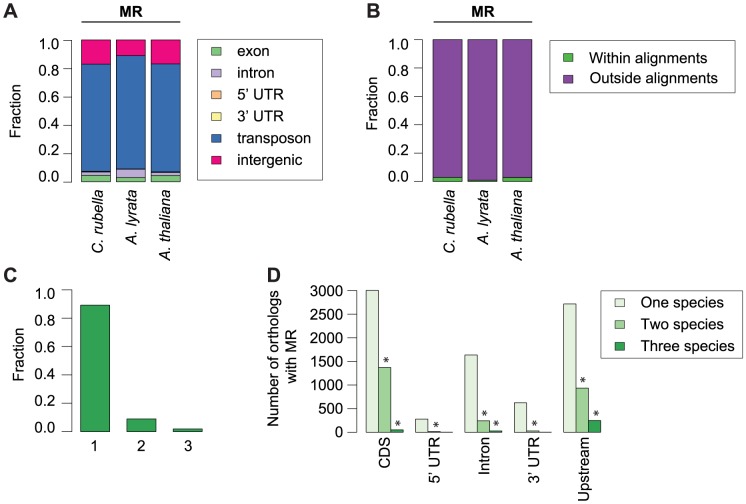
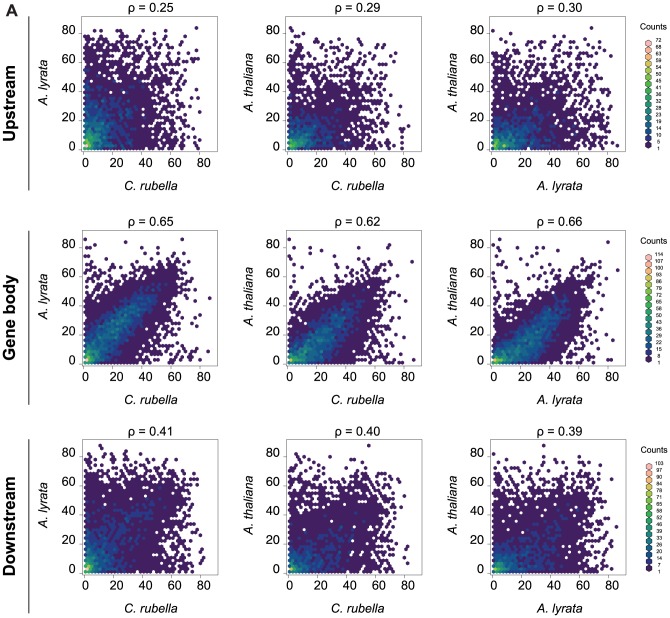
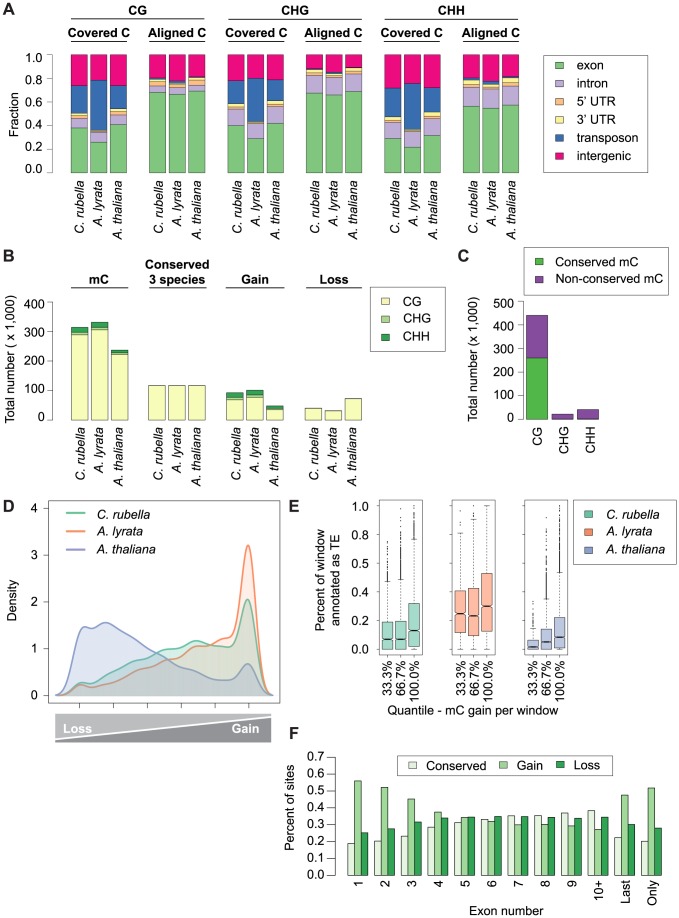
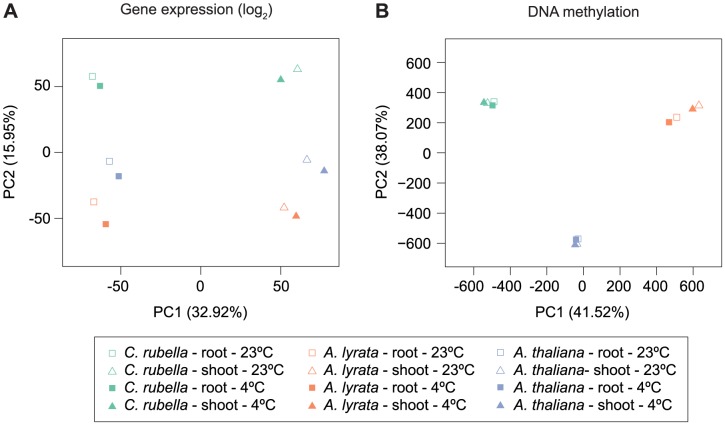
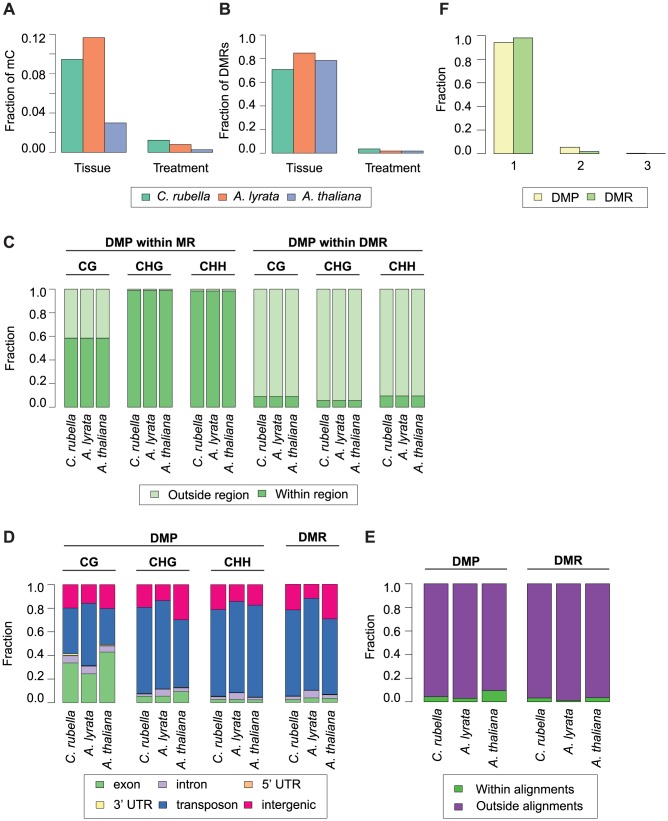
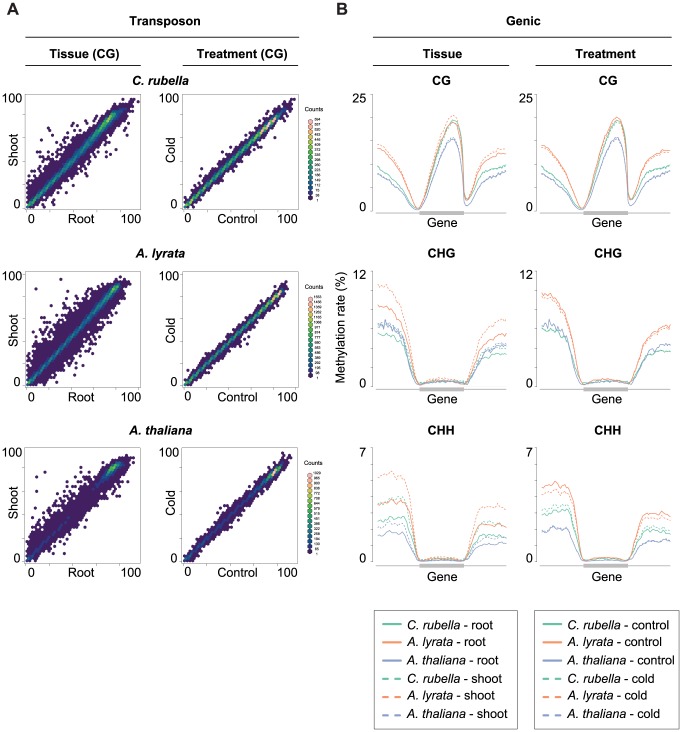
DNA methylation is an ancient molecular modification found in most eukaryotes. In plants, DNA methylation is not only critical for transcriptionally silencing transposons, but can also affect phenotype by altering expression of protein coding genes. The extent of its contribution to phenotypic diversity over evolutionary time is, however, unclear, because of limited stability of epialleles that are not linked to DNA mutations. To dissect the relative contribution of DNA methylation to transposon surveillance and host gene regulation, we leveraged information from three species in the Brassicaceae that vary in genome architecture, Capsella rubella, Arabidopsis lyrata, and Arabidopsis thaliana. We found that the lineage-specific expansion and contraction of transposon and repeat sequences is the main driver of interspecific differences in DNA methylation. The most heavily methylated portions of the genome are thus not conserved at the sequence level. Outside of repeat-associated methylation, there is a surprising degree of conservation in methylation at single nucleotides located in gene bodies. Finally, dynamic DNA methylation is affected more by tissue type than by environmental differences in all species, but these responses are not conserved. The majority of DNA methylation variation between species resides in hypervariable genomic regions, and thus, in the context of macroevolution, is of limited phenotypic consequence.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Zemach A, McDaniel IE, Silva P, Zilberman D (2010) Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science 328: 916–919. - PubMed
-
- Suzuki MM, Bird A (2008) DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet 9: 465–476. - PubMed
-
- Gruenbaum Y, Navehmany T, Cedar H, Razin A (1981) Sequence specificity of methylation in higher-plant DNA. Nature 292: 860–862. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
