

Oligomeric Aβ-induced synaptic dysfunction in Alzheimer's disease
- PMID: 25394486
- PMCID: PMC4237769
- DOI: 10.1186/1750-1326-9-48
Oligomeric Aβ-induced synaptic dysfunction in Alzheimer's disease
Abstract
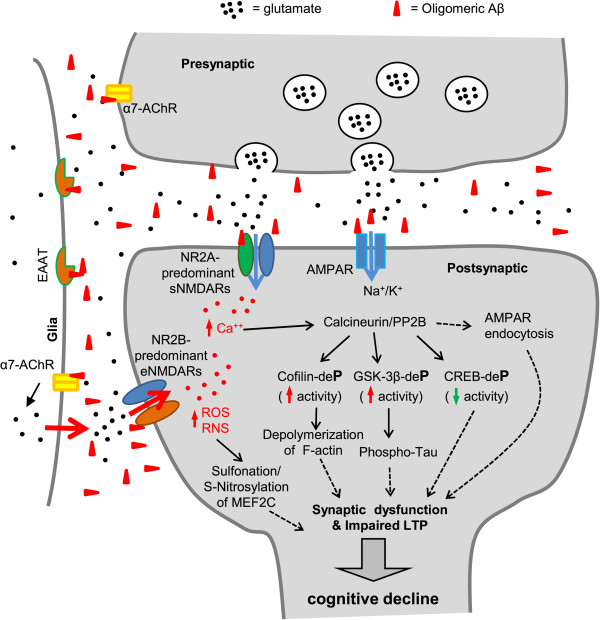
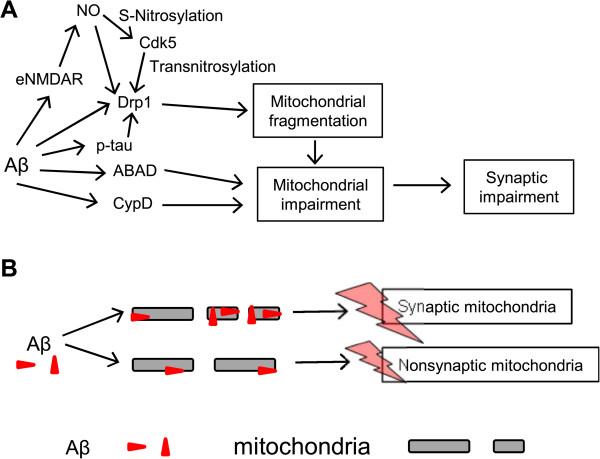
Alzheimer's disease (AD) is a devastating disease characterized by synaptic and neuronal loss in the elderly. Compelling evidence suggests that soluble amyloid-β peptide (Aβ) oligomers induce synaptic loss in AD. Aβ-induced synaptic dysfunction is dependent on overstimulation of N-methyl-D-aspartate receptors (NMDARs) resulting in aberrant activation of redox-mediated events as well as elevation of cytoplasmic Ca2+, which in turn triggers downstream pathways involving phospho-tau (p-tau), caspases, Cdk5/dynamin-related protein 1 (Drp1), calcineurin/PP2B, PP2A, Gsk-3β, Fyn, cofilin, and CaMKII and causes endocytosis of AMPA receptors (AMPARs) as well as NMDARs. Dysfunction in these pathways leads to mitochondrial dysfunction, bioenergetic compromise and consequent synaptic dysfunction and loss, impaired long-term potentiation (LTP), and cognitive decline. Evidence also suggests that Aβ may, at least in part, mediate these events by causing an aberrant rise in extrasynaptic glutamate levels by inhibiting glutamate uptake or triggering glutamate release from glial cells. Consequent extrasynaptic NMDAR (eNMDAR) overstimulation then results in synaptic dysfunction via the aforementioned pathways. Consistent with this model of Aβ-induced synaptic loss, Aβ synaptic toxicity can be partially ameliorated by the NMDAR antagonists (such as memantine and NitroMemantine). PSD-95, an important scaffolding protein that regulates synaptic distribution and activity of both NMDA and AMPA receptors, is also functionally disrupted by Aβ. PSD-95 dysregulation is likely an important intermediate step in the pathological cascade of events caused by Aβ. In summary, Aβ-induced synaptic dysfunction is a complicated process involving multiple pathways, components and biological events, and their underlying mechanisms, albeit as yet incompletely understood, may offer hope for new therapeutic avenues.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
- R21 NS080799/NS/NINDS NIH HHS/United States
- R01 AG038710/AG/NIA NIH HHS/United States
- R01AG021173/AG/NIA NIH HHS/United States
- R01 AG021173/AG/NIA NIH HHS/United States
- P30 NS076411/NS/NINDS NIH HHS/United States
- R01AG038710/AG/NIA NIH HHS/United States
- R01 AG044420/AG/NIA NIH HHS/United States
- R01NS046673/NS/NINDS NIH HHS/United States
- R01 NS086890/NS/NINDS NIH HHS/United States
- P01 HD029587/HD/NICHD NIH HHS/United States
- R01 NS046673/NS/NINDS NIH HHS/United States
- R21 MH102672/MH/NIMH NIH HHS/United States
- P01 HD29587/HD/NICHD NIH HHS/United States
- R01AG044420/AG/NIA NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
