

Truncating mutations in the last exon of NOTCH3 cause lateral meningocele syndrome
- PMID: 25394726
- PMCID: PMC5589071
- DOI: 10.1002/ajmg.a.36863
Truncating mutations in the last exon of NOTCH3 cause lateral meningocele syndrome
Abstract
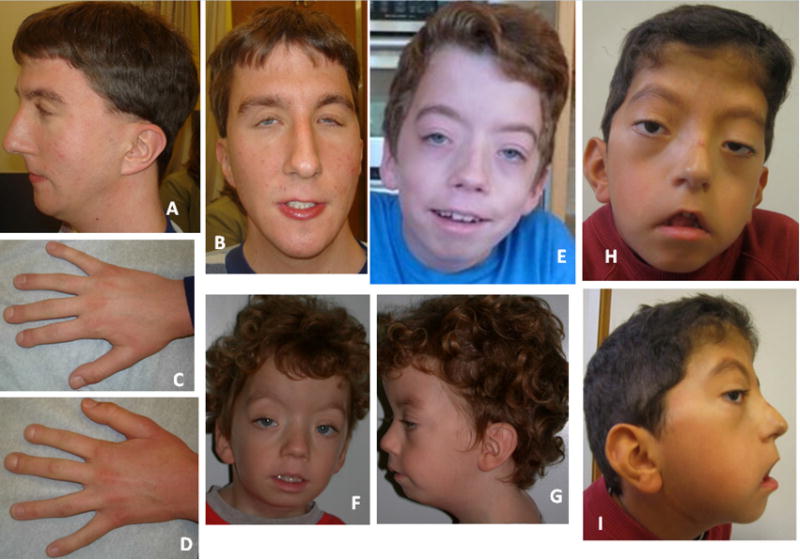
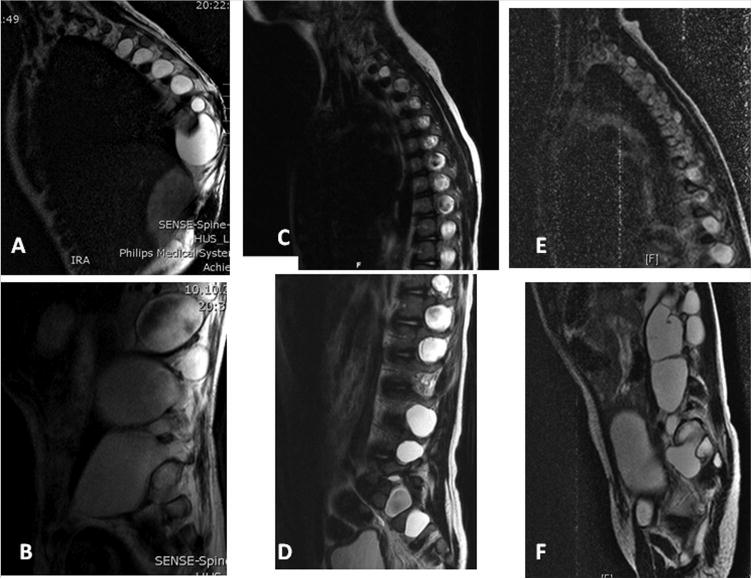
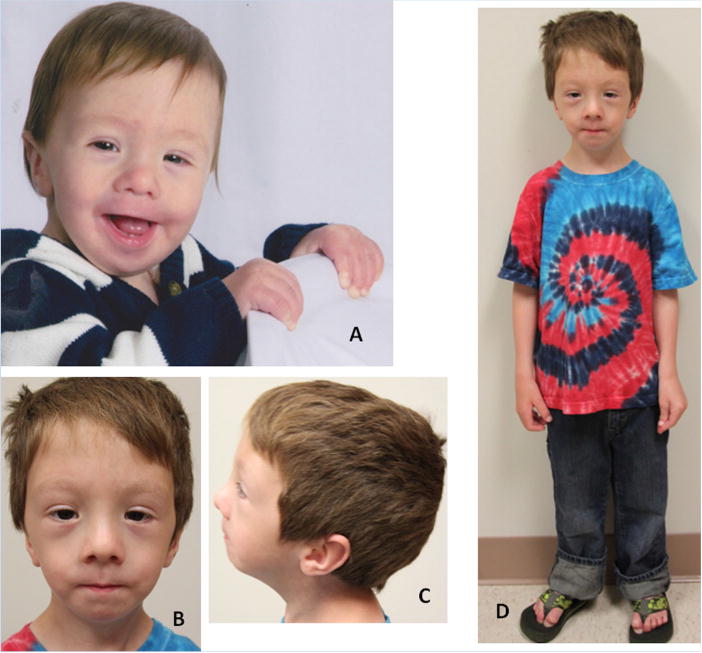
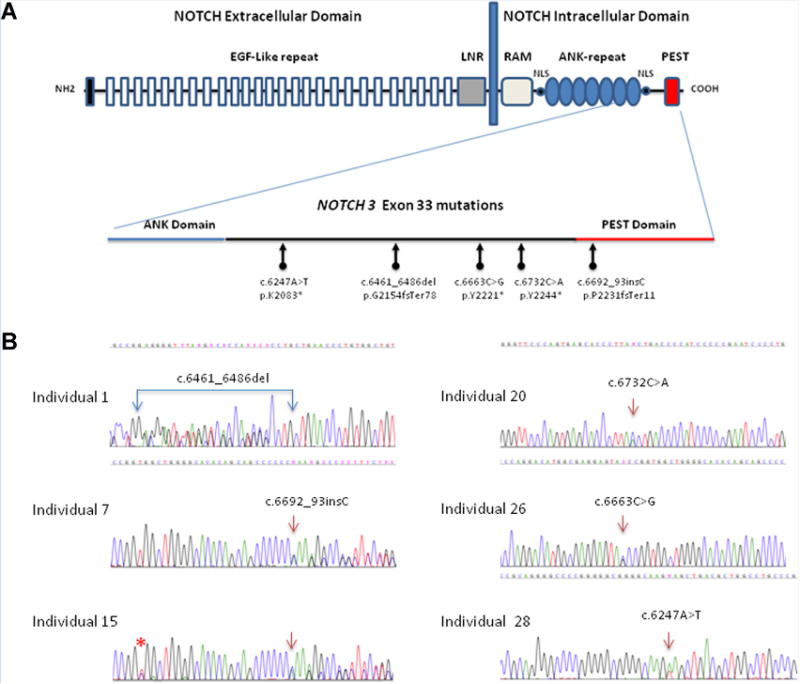
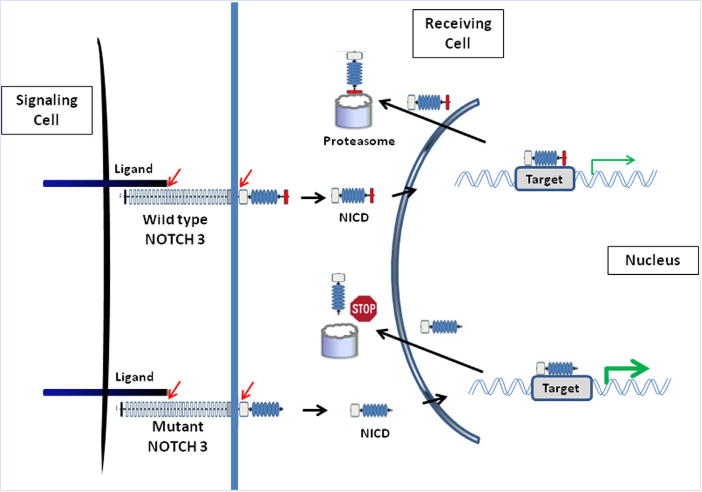
Lateral meningocele syndrome (LMS, OMIM%130720), also known as Lehman syndrome, is a very rare skeletal disorder with facial anomalies, hypotonia and meningocele-related neurologic dysfunction. The characteristic lateral meningoceles represent the severe end of the dural ectasia spectrum and are typically most severe in the lower spine. Facial features of LMS include hypertelorism and telecanthus, high arched eyebrows, ptosis, midfacial hypoplasia, micrognathia, high and narrow palate, low-set ears and a hypotonic appearance. Hyperextensibility, hernias and scoliosis reflect a connective tissue abnormality, and aortic dilation, a high-pitched nasal voice, wormian bones and osteolysis may be present. Lateral meningocele syndrome has phenotypic overlap with Hajdu-Cheney syndrome. We performed exome resequencing in five unrelated individuals with LMS and identified heterozygous truncating NOTCH3 mutations. In an additional unrelated individual Sanger sequencing revealed a deleterious variant in the same exon 33. In total, five novel de novo NOTCH3 mutations were identified in six unrelated patients. One had a 26 bp deletion (c.6461_6486del, p.G2154fsTer78), two carried the same single base pair insertion (c.6692_93insC, p.P2231fsTer11), and three individuals had a nonsense point mutation at c.6247A > T (pK2083*), c.6663C > G (p.Y2221*) or c.6732C > A, (p.Y2244*). All mutations cluster into the last coding exon, resulting in premature termination of the protein and truncation of the negative regulatory proline-glutamate-serine-threonine rich PEST domain. Our results suggest that mutant mRNA products escape nonsense mediated decay. The truncated NOTCH3 may cause gain-of-function through decreased clearance of the active intracellular product, resembling NOTCH2 mutations in the clinically related Hajdu-Cheney syndrome and contrasting the NOTCH3 missense mutations causing CADASIL.
Keywords: Hajdu-Cheney syndrome; Lehman syndrome; NOTCH3; PEST domain; dural ectasia; lateral meningocele syndrome.
© 2014 Wiley Periodicals, Inc.
Conflict of interest statement
Conflict of interest: none
Figures
References
-
- Avela K, Makitie O. Response to “Lateral Meningocele Syndrome and Hajdu–Cheney syndrome: Different disorders with overlapping phenotypes” by Gripp. Am J Med Genet Part A. 2011;155A:1775. - PubMed
-
- Avela K, Valanne L, Helenius I, Makitie O. Hajdu–Cheney syndrome with severe dural ectasia. Am J Med Genet Part A. 2011;155A:595–598. - PubMed
-
- Bellavia D, Checquolo S, Campese AF, Felli MP, Gulino A, Screpanti I. Notch3: From subtle structural differences to functional diversity. Oncogene. 2008;27:5092–5098. - PubMed
-
- Castori M, Morlino S, Ritelli M, Brancati F, de Bernardo C, Colombi M, Grammatico P. Late diagnosis of lateral meningocele syndrome in 55-year-old woman with symptoms of joint instability and chronic musculoskeletal pain. Am J Med Genet Part A. 2014;164A:528–534. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
